PSU Volume 57 NO 01 JULY 2021
Neonatal Gastrostomy
One of the main reasons for prolonged hospitalization in
newborn infants is delay in achieving full oral feedings. Infants who
are unable to orally feed or have insufficient oral intakes require
longer hospitalization after birth. Prematurity impacts negatively in
the attainment of feeding milestones and 40% of infants referred to
feeding clinics are born preterm. Infants may not be able to achieve
full oral feedings due to congenital anomalies, being congenital heart
disease one of the main reason, genetic conditions, neurologic injury,
respiratory insufficiency and extreme prematurity. Feeding difficulty
in infants in the neonatal period are related to sensory or motor
neurologic vulnerabilities, static or progressive neurological disease,
behavioral deficits, chronic lung disease, gastrointestinal disorders
or a combination of all of these etiologies. When the neonate cannot
consume adequate oral feeding, is gavage-tube feeding dependent,
suffers from postprandial related cardio-respiratory spells, refuse to
feed or has poor sucking ability a gastrostomy tube placement is
performed. Gastrostomy placement is most strongly associated with
bronchopulmonary dysplasia, intraventricular hemorrhage or
periventricular leukomalacia and small for gestational age status.
Gavages tubes when dislodge results in a high choking and
aspiration risk, are associated with leaks, infection, reflux and
feeding aversion. Nevertheless, home nasogastric feeding is a
particular good alternative in infants discharge home on room air or
nasal cannulas who are taking almost 50% of feeds orally.Gastrostomy
tube are either placed open, laparoscopically or using endoscopic
technique (PEG) depending on the preference of the surgeon. Gastrostomy
tubes are placed when the baby attains at least 3 Kg of weight.
Surgeons are moving to laparoscopic gastrostomy as the standard of
care. Carbon dioxide insufflation and absorption during short
laparoscopic procedures have demonstrated no significant alteration in
cerebral or renal oxygenation or oxygen extraction. Premature infants
that undergo gastrostomy placement have a significant increased risk of
both inpatient readmission and emergency department visits within three
months of NICU discharge. One-third of infants with g-tube have at
least one emergency department visit and 9% multiple, with inadvertent
removal/misplacement of the tube being the most common cause. For NICU
infants who cannot feed or take medications by mouth, gastrostomy tube
represents a safe way of administering nutrition and medications for
long periods of time. After placement of the g-tube, infants may take
more than two weeks to gain weight at rates seen prior to the surgery.
Weight gain and appropriate growth occur more frequently in the
population of children with neurodevelopmental disability when g-tubes
placement occurs early, before morbidity and malnutrition become
evident. Additional benefits of g-tube placement in newborns include
safety of administration of nutrients, fluids, and medications, as well
as facilitating discharge planning, including parental comfort and
decreased stress regarding the long-term nutritional status of the
baby.
References:
1- Jadcherla SR, Khot T, Moore R, Malkar M, Gulati IK, Slaughter JL:
Feeding Methods at Discharge Predict Long-Term Feeding and
Neurodevelopmental Outcomes in Preterm Infants Referred for Gastrostomy
Evaluation. J Pediatr. 181: 125-130, 2017
2- Duncan TL, Ulugia J, Bucher BT: Association of gastrostomy placement
on hospital readmission in premature infants. J Perinatol.
39(11):1485-1491, 2019
3- Munoz A, Tan J, Hopper A, Vannix R, Carter H, Woodfin M, Blood A,
Baerg J: Cerebral and Renal Oxygenation in Infants Undergoing
Laparoscopic Gastrostomy Tube Placement. J Surg Res. 256:83-89, 2020
4- Puia-Dumitrescu M, Benjamin DK Sr, Smith PB, et al: Impact of
Gastrostomy Tube Placement on Short-Term Weight Gain in Hospitalized
Premature Infants. JPEN J Parenter Enteral Nutr. 44(2):355-360, 2020
5- Chapman A, George K, Selassie A, Lesher AP, Ryan RM: NICU infants
who require a feeding gastrostomy for discharge. J Pediatr Surg.
56(3):449-453, 2021
6- Khalil ST, Uhing MR, Duesing L, et al: Outcomes of Infants With Home
Tube Feeding: Comparing Nasogastric vs Gastrostomy Tubes. JPEN J
Parenter Enteral Nutr. 41(8):1380-1385, 2017
7- Williams SL, Popowics NM, Tadesse DG, Poindexter BB, Merhar SL: Tube
feeding outcomes of infants in a Level IV NICU. J Perinatol.
39(10):1406-1410, 2019
Multiple Endocrine Neoplasia Type 2
The multiple endocrine neoplasia type 2 (MEN-2) is a rare
autosomal dominant inherited disorder caused by a germline mutation in
the RET protooncogene with a prevalence of one in 30,000 live births.
The RET protooncogene encodes the transmembrane tyrosine kinase
receptor on chromosome 10q11.2. MEN-2 consists of three different
syndromes: the MEN-2A (80-90%) characterized by medullary thyroid
carcinoma (MTC), pheochromocytoma and primary hyperparathyroidism, the
MEN-2B (5-10%) characterized by MTC, pheochromocytoma in children with
distinct physical manifestation such as marfanoid habitus and multiple
neuromas, and the familial MTC. Direct DNA analysis allows
identification of children with MEN-2A. MTC is usually the first
neoplasm to develop in 90-100% of cases, and the most common cause of
death in MEN patients. This is the reason why prophylactic total
thyroidectomy before the age of 5 years is recommended to those with
the RET protooncogene mutation of codon 634 in the extracellular domain
of the receptor. MTC in MEN-2 children can be cured by surgically
removing all the c-cells at risk of becoming malignant. Calcitonin is
utilized as marker of residual or recurrent disease. Pheochromocytomas
has an incidence of 50% in MEN-2 syndromes, they are diagnosed at an
earlier age, mostly of adrenal origin, rarely becomes metastatic,
although they most almost always develop bilaterally. Management is
surgical excision of the tumor harboring the pheochromocytoma. Cortical
sparing adrenalectomy can be performed as part of bilateral adrenal
resection. Hyperparathyroidism caused by hyperplasia of the gland
occurs in 35% of patients with MEN-2. A group of children with MEN-2A
develop Hirschsprung's disease (HD). Diagnosis is through rectal biopsy
and management is pull-through surgery. In MEN-2B, pheochromocytomas
develop in 50% of patients and all patients have neural gangliomas,
particularly in the mucosa of the digestive tract, conjunctiva, lips
and tongue. MEN-2B do not develop hyperparathyroidism. MTC in MEN-2B
develop at a very young age (infancy) and appears to be the most
aggressive form of hereditary MTC. Prophylactic total thyroidectomy is
recommended before the age of two years in children with MEN-2B.
Gastrointestinal ganglioneuromatosis is the predominant etiology of
most alimentary tract symptoms in children with MEN-2B, resulting in
thickening of myenteric plexus and ganglion cell hypertrophy leading to
loss of normal bowel tone, distension, segmental dilatation and
megacolon, though the number of ganglion cells is not reduced or absent
as with HD. They develop constipation and intermittent diarrhea.
Management is conservative, as symptoms are less severe than MEN-2A
with HD. Marfanoid habitus is present in 65-75% of children with MEN-2B
characterized by elongated face, large hands and feet with relatively
long extremities. Skeletal anomalies include lordosis, kyphosis, joint
laxity and talipes equino varus. 86% of MEN-2B are unable to shed
tears. Familial MTC represent the remaining hereditary MTC cases and is
characterized by presence of MTC without pheochromocytoma,
hyperparathyroidism or physical characteristics of MEN-2B. MTC has a
late onset with a good prognosis in the majority of familial cases.
Late genetic testing, surgery beyond recommended age and elevated basal
calcitonin levels are factors associated with higher rate of MTC in the
specimen. No lymph node metastasis is present with basal calcitonin
below 40 pg/ml. Above that level or in the presence of clinical
palpable lymph nodes central lymph node dissection is recommended
during thyroidectomy.
References:
1- Danko ME, Skinner MA: Surgical intervention in children with
multiple endocrine neoplasia type 2. Curr Opin Pediatr. 18(3):312-5,
2006
2- Martucciello G, Lerone M, Bricco L, Tonini GP, Lombardi L, Del Rossi
CG, Bernasconi S: Multiple endocrine neoplasias type 2B and RET
proto-oncogene. Ital J Pediatr. 38:9, 2012
3- Machens A, Dralle H: Multiple endocrine neoplasia type 2:
achievements and current challenges. Clinics (Sao Paulo). 67 Suppl
1(Suppl 1):113-8, 2012
4- Wohllk N, Schweizer H, Erlic Z, et al: Multiple endocrine neoplasia
type 2. Best Pract Res Clin Endocrinol Metab. 24(3):371-87, 2010
5- Prete FP, Abdel-Aziz T, Morkane C, et al: Prophylactic thyroidectomy
in children with multiple endocrine neoplasia type 2. Br J Surg.
105(10):1319-1327, 2018
6- Bussieres V, Roy S, Deladoey J, et al: Prophylactic thyroidectomies
in MEN2 syndrome: Management and outcomes. J Pediatr Surg. 53: 283-285,
2018
7- Ordonez J, Perez-Egido L, Garcia-Casillas, et al: Management and
results of thyroidectomies in pediatric patients with MEN 2 syndrome. J
Pediatr Surg. https://doi.org/10.1016/j.pedsurg.2021.02.061
Functioning Adrenocortical Tumors
Adrenocortical tumors (ACT) in children are rare, comprising
10-25% of all adrenal neoplasms. It is estimated that 25 new cases are
seen each year in the US. The incidence is high in southern Brazil due
to a high rate of mutation in the tumor suppression gene p53. Most of
these tumors (95%) are functional producing hormones such as androgens,
cortisol, aldosterone and estrogens in decreasing order of frequency.
In cases who present with virilization, the most prominent symptom is
rapid growth, acne, deepening of the voice, advanced bone age,
clitoromegaly or penile enlargement. Functional adrenocortical tumors
have a good prognosis when managed appropriately. Functioning ACT has a
peak presentation during the first decade of life and occur more
commonly in females. A family history is common in cases of ACT.
Survival rates are better in children with ACT than adults. ACT in
children are associated with Beckwith-Weidman, MEN-1, Carney complex,
congenital adrenal hyperplasia or Li-Fraumeni syndromes. The most
common clinical presentation is virilization, followed by cushingoid
features, hypertension, hyperestrogenism or a combination of these
clinical manifestations. Adrenocortical adenomas and carcinomas can
occur both in children with ACT. Presence of metastasis is absolute
evidence of malignancy. Criteria suggesting malignancy include large
tumor size, tumor weight exceeding 400 gm, extension into periadrenal
soft tissue, high nuclear grade, high mitotic rate per high power
field (> 15 mitotic figure per 20 HPF), atypical mitosis, diffuse
architecture, necrosis, capsular invasion and vascular invasion. ACT in
young children and infants are more likely associated with the best
overall prognosis and may not be as uniformly fatal as they are in
older children. A thorough hormonal evaluation is needed for a precise
classification of functioning ACT even if there is no clinical sign or
symptom of hormone excess. Most ACT are located in the left adrenal
gland. Most imaging modalities (US, CT and MRI) can detect the adrenal
tumor. Management of ACT is surgical excision of the affected adrenal
gland. The laparoscopic approach for removing the adrenal gland is the
gold standard in all functioning ACT except the adrenocortical
carcinoma, since minimal tumor spillage changes negatively the
prognosis dramatically. In the postoperative follow-up, positron
emission tomography with computer tomography (PET-CT) can be helpful in
the detection of secondary lesions. Cryoablation should be considered
in rare selected cases of tumors that are not amenable to surgical
resection.
References:
1- Patil KK, Ransley PG, McCullagh M, Malone M, Spitz L: Functioning
adrenocortical neoplasms in children. BJU Int. 89(6):562-5, 2002
2- Ahmed AA: Adrenocortical neoplasms in young children: age as a prognostic factor. Ann Clin Lab Sci. 39(3):277-82, 2009
3- Ghazi AA, Mofid D, Salehian MT, et al: Functioning adrenocortical
tumors in children-secretory behavior. J Clin Res Pediatr Endocrinol.
5(1):27-32, 2013
4- Mihai R: Rare adrenal tumors in children. Semin Pediatr Surg. 23(2):71-5, 2014
5- Gupta N, Rivera M, Novotny P, Rodriguez V, Bancos I, Lteif A:
Adrenocortical Carcinoma in Children: A Clinicopathological Analysis of
41 Patients at the Mayo Clinic from 1950 to 2017. Horm Res Paediatr.
90(1):8-18, 2018
6- Lopes RI, Suartz CV, Neto RP, et al: Management of functioning pediatric adrenal tumors. J Pediatr Surg. 56: 768-771, 2021
PSU Volume 57 NO 02 AUGUST 2021
Mucous Fistula Refeeding
Neonates require enterostomy for a variety of conditions
such as congenital atresias, meconium ileus, midgut volvulus,
necrotizing enterocolitis, spontaneous bowel perforation and rarely
gastroschisis. Substantial surgical resection is associated with a
short residual length of bowel, often with a proximal stoma and distal
mucous fistula. Stomas located in the jejunum or proximal ileum are
classified as high output stomas resulting in production of large
ostomy losses, fluid and electrolytes imbalances, metabolic acidosis,
impaired absorption of fat, protein and other nutrients. In neonates
with jejunal enterostomy and mucous fistula a significant length of
bowel may be defunctionalized and not be able to be used for absorption
of nutrients and electrolytes. Though they anatomically have near
normal length of bowel, the higher the enterostomy, the higher the
complications associated with a functional small bowel syndrome. These
infants rely on total parenteral nutrition (TPN) for growth and
development. Mucous fistula refeeding (MFR) is the practice of
collecting proximal ostomy effluent and reinfusing it into the distal
mucous fistula. Refeeding the distal defunctionalized small bowel
through the mucous fistula using the proximal succus entericus
secretions can reduce the complications associated with a short bowel
syndrome. The clinical benefits of MFR include simplified control of
fluids and electrolytes balance in patients with high stoma output,
optimal utilization of the remaining absorptive capacity for enteral
nutrition, and reduction of gastrointestinal proximal stoma secretions
up to 30%. MFR can be used with and without TPN preventing the atrophy
of the distal bowel while preparing it for reanastomosis. Refeeding the
proximal stoma effluent through the distal mucous fistula uses the
absorptive surface of the distal bowel for nutrient absorption,
stimulates mucosal growth and intestinal adaptation and prevents
atrophy of the villi of the defunctionalizedl bowel. The increase
absorptive function from the added length of intestine may reduce the
requirement for parenteral nutrition, promote better weight gain and
help eliminate cholestasis by stimulating the enterohepatic
circulation. The aim of the MFR technique in infants who have
undergone bowel resection is to prime the bowel with luminal feeding
promoting intestinal adaptation such as cell hyperplasia, bowel
hypertrophy, lengthening and heightening of villi, improved peristalsis
and mucosal growth. Strong intestinal growth stimulants including
peptides and nutrient substances present in high concentration in the
proximal enterostomy effluent induce substantial bowel lengthening and
hypertrophy. Disuse atrophy of distal loop can be prevented. A further
advantage of the MFR technique is simplification of the control of
fluids and electrolytes balance in neonates with a high stoma that has
a large output. Indications for refeeding of stoma effluent into the
mucous fistula include the presence of a proximal stoma, a high-output
enterostomy, electrolytes disturbance or failure to achieve adequate
weight gain. Prior to initiation of MFR, patency of the distal bowel is
ensured by means of a contrast fluoroscopy study through the mucous
fistula. Infuse rate should not exceed 6-10 ml/hr with output refeeding
performed every 3 hours to avoid bacterial overgrowth of the effluent
to be used. Enteral refeeding technique is safe, reduce hospital stay,
improves weight gain and potentially reduces TPN use and related
complications in infants with small bowel syndrome and high output
enterostomies. Complications associated with MFR include bowel
perforation with the use of the catheter, bleeding, bacterial
overgrowth if there is a delay between collection and refeeding of the
stoma effluent.
References:
1- Gardner VA, Walton JM, Chessell L: A case study utilizing an enteral
refeeding technique in a premature infant with short bowel syndrome.
Adv Neonatal Care. 3(6):258-68, 2003
2- Richardson L, Banerjee S, Rabe H: What is the evidence on the
practice of mucous fistula refeeding in neonates with short bowel
syndrome? J Pediatr Gastroenterol Nutr. 43(2):267-70, 2006
3- Haddock CA, Stanger JD, Albersheim SG, Casey LM, Butterworth SA:
Mucous fistula refeeding in neonates with enterostomies. J Pediatr
Surg. 50(5):779-82, 2015
4- Lau EC, Fung AC, Wong KK, Tam PK: Beneficial effects of mucous
fistula refeeding in necrotizing enterocolitis neonates with
enterostomies. J Pediatr Surg. 51(12):1914-1916, 2016
5- Gause CD, Hayashi M, Haney C, et al: Mucous fistula refeeding
decreases parenteral nutrition exposure in postsurgical premature
neonates. J Pediatr Surg. 51(11):1759-1765, 2016
6- Elliott T, Walton JM: Safety of mucous fistula refeeding in neonates with functional short bowel
syndrome: A retrospective review. J Pediatr Surg. 54(5):989-992, 2019
7- Yabe K, Kouchi K, Takenouchi A, Matsuoka A, Korai T, Nakata C:
Safety and efficacy of mucous fistula refeeding in low-birth-weight
infants with enterostomies. Pediatr Surg Int. 35(10):1101-1107, 2019
8- Ghattaura H, Borooah M, Jester I: A Review on Safety and Outcomes of
Mucous Fistula Refeeding in Neonates. Eur J Pediatr Surg. 2020 Nov 10.
doi: 10.1055/s-0040-1718751.
Atypical Mycobacterias
Atypical mycobacteria infection refers to disease produce by
Nontuberculous mycobacteria (NTM). They are environmental
acid-fast organisms isolated from soil, water, milk, eggs, vegetables
and animals transmitted to humans through the respiratory system. More
than 130 species have been identified, many of which cause human
disease. In children, infection with NTM can result in cervical
lymphadenitis, skin and osteoarticular infections, lung disease
(predominantly in children with chronic lung disease), and disseminated
disease infection in immune-compromised children. Mycobacterium
Avium-Intracellulare complex (MAC) is usually the most frequently NTM
isolated in children. Cystic fibrosis and Mendelian susceptibility to
mycobacterial disease are two distinct inborn genetic disorders
associated with NTM disease. Of the acquired disorders associated with
NTM, HIV infection predominates. The most common NTM-associated disease
in healthy children is chronic cervicofacial lymphadenitis most
frequently caused by mycobacterium avium/Intracellulare complex. The
oropharyngeal mucosa is the typical portal of entry as toddlers place
contaminated objects in their mouths. NTM lymphadenitis occur early in
childhood with 80% in children younger than five years, with a mean age
of diagnosis of 2.5 years. Children present with history of unilateral
lymph node swelling usually affecting the jugulodigastric, parotid or
preauricular, submandibular and posterior triangle lymph node
persisting for weeks to months despite antibiotic therapy. The
infection is not associated with systemic symptoms or signs.
Involvement of submandibular lymph nodes represents the most frequent
localization (80%). The affected lymph node can go from a painless firm
mass with increased vascularity, to a more fluctuant mass due to
liquefaction. Next the skin over the lymph node takes a violaceous
discoloration which might lead to fistulization that may discharge for
months. Spontaneous healing usually occurs within six months. Pulmonary
NTM disease is indistinguishable from pulmonary tuberculosis and is
usually associated with cystic fibrosis. Diagnosis of NTM disease
requires clinical, radiological and microbiological assessment. A
tuberculin (PPD) induration greater than 5 mm at 48 hours suggest a
diagnosis of NTM infection. Microbiological diagnosis of NTM disease is
achieved by detection of the causative organisms by PCR (more
sensitive; more rapid), or bacterial culture (slow growth). Molecular
detection of NTM in lymph node biopsy samples is more sensitive than
bacterial culture. Histopathology reveals necrotizing granulomatous
inflammation associated with caseous necrotic areas. Interferon gamma
release assay (IGRA) is positive in 70-80% of tuberculosis lymphangitis
cases and generally negative in NTM. The characteristic radiological
feature of NTM infection is the presence of central cavitating lesions
represented by low-density necrotic material. Management of NTM disease
relies on combination of several antibiotics, with macrolide being the
cornerstone of treatment. Treatment of NTM adenitis depends on disease
stage and severity. Lack of response to three months of antibiotic
therapy is considered a treatment failure. Surgery remains an option
for lesions that show evidence of progression to cutaneous
involvement. Complete surgical excision of the affected lymph
node, as soon as possible, is regarded as the best curative option.
Secondary wound infection and permanent injury to the facial nerve is a
major concern with surgical excision of affected lymph nodes. In cases
of incomplete excision of the infected lymph node a
macrolide-containing drug regimen should be given. Fluctuant lesions
are managed more frequently with antibiotics, while a firm lesion can
be observed for spontaneous resolution.
References:
1- Lopez-Varela E, Garcia-Basteiro AL, Santiago B, Wagner D, van Ingen
J, Kampmann B: Non-tuberculous mycobacteria in children: muddying the
waters of tuberculosis diagnosis. Lancet Respir Med. 3(3):244-56, 2015
2- Naselli A, Losurdo G, Avanzini S, et al: Management of
nontuberculous mycobacerial lymphadenitis in a tertiary care children's
hospital: A 20 year experience. J pediatr Surg. 52: 593-597, 2017
3- Loizos A, Soteriades ES, Pieridou D, Koliou MG: Lymphadenitis by
non-tuberculous mycobacteria in children. Pediatr Int.
60(12):1062-1067, 2018
4- Torretta S, Gaffuri M, Ibba T, et al: Surgical treatment of
non-tuberculous mycobacterial lymphadenitis in children: Our experience
and a narrative review. Int J Immunopathol Pharmacol. 2018
Jan-Dec;32:2058738418806413. doi:10.1177/2058738418806413.
5- Gallois Y, Cogo H, Debuisson C, et al: Nontuberculous lymphadenitis
in children: What management strategy? Int J Pediatr Otorhinolaryngol.
22:196-202, 2019
6- Meoli A, Deolmi M, Iannarella R, Esposito S: Non-Tuberculous Mycobacterial Diseases in Children. Pathogens. 9(7):553, 2020
7- Lyly A, Kontturi A, Salo E, Nieminen T, Nokso-Koivisto J: Childhood
nontuberculous mycobacterial lymphadenitis-observation alone is a good
alternative to surgery. Int J Pediatr Otorhinolaryngol. 2020
Feb;129:109778. doi:10.1016/j.ijporl.2019.109778.
Accessory Cardiac Bronchus
Accessory cardiac bronchus (ACB) is a very rare, poorly
recognized, usually asymptomatic congenital anomaly of the
tracheo-bronchial tree. ACB is a supernumerary bronchus usually arising
from the inner wall of the right main or intermediate bronchus,
opposite to the origin of the right upper lobe bronchus. Most cases are
incidental findings in asymptomatic adult patients. ACB is a true
bronchus, with normal epithelial lining and cartilage walls. The ACB is
thought to be a remnant of the cardiac bronchial bud that failed to
regress during embryogenesis. Three anatomic variations of ACB have
been described: a short, blind ending type, an accessory lobed type
which branches into rudimentary ventilated lobules, and a long
diverticular type lacking any further arborization. The configuration
may range from a short diverticulum where no lung tissue is observed
and it appears as a stump, to a longer structure where surrounding lung
tissue is present. 70% of ACB are of the diverticulum type ending
blindly. Usually, ACB arises from the medial wall of the bronchus
intermedius (75%), has a mean diameter of 8.7 mm and a mean length of
12 mm. It is lined by a normal bronchial mucosa, has cartilage within
its wall and is usually demarcated by a spur at its origin from the
normal bronchus. Though most cases are asymptomatic, ACB may be a site
of chronic inflammation and produce several complications including
recurrent secondary lung infection, hemoptysis, chronic cough and
rarely malignant transformation. Diagnosis is established with chest
CT-Scan. Bronchoscopy might miss the accessory bronchus due to
constriction by repeated inflammation. The recognition of an ACB is
important since it should be differentiated from acquired bronchial
fistula, diverticulum or adenoid recess. Surgical excision of ACB is
recommended when symptomatic, or in asymptomatic patients with the
lobed or long diverticular type because of the high probability of
long-term complications. This can be accomplished using either minimal
invasive thoracoscopy or open thoracotomy.
References:
1- White ES: Accessory cardiac bronchus. Am J Respir Crit Care Med. 183(6):825, 2011
2- Barreiro TJ, Gemmel D: Accessory cardiac bronchus. Lung. 192(5):821-2, 2014
3- Volpe A, Bozzetto S, Baraldi E, Gamba P: Accessory-lobed accessory
cardiac bronchus: Presentation and treatment in a pediatric patient.
Pediatr Pulmonol. 52(10):E85-E87, 2017
4- Ghaye B, Collard P, Pierard S, Sluysmans T: CT presentation of
left-sided accessory cardiac bronchus. Diagn Interv Imaging.
99(12):827-828, 2018
5- Wong LM, Cheruiyot I, Santos de Oliveira MH, et al: Congenital
anomalies of the tracheobronchial tree: a meta-analysis and clinical
considerations. Ann Thorac Surg. 2020 Nov 4:S0003-4975(20)31854-3. doi:
10.1016/j.athoracsur.2020.08.060.
6- Yildiz H, Ugurel S, Soylu K, Tasar M, Somuncu I: Accessory cardiac
bronchus and tracheal bronchus anomalies: CT-bronchoscopy and
CT-bronchography findings. Surg Radiol Anat. 28(6):646-9, 2006
PSU Volume 57 NO 03 SEPTEMBER 2021
Androgen Insensitivity Syndrome
Complete virilization of a 46XY fetus depends on either
androgens or a functioning androgen receptor. Androgen insensitivity
syndrome (AIS) is an X-linked recessive genetic condition caused by an
androgen receptor gene mutation situated in the Xq11-q12 region, which
results in resistance to androgens in 46XY individuals. The disorder is
characterized by the presence of a male karyotype with a female
phenotype. AIS is divided into subtypes that include complete AIS
(complete feminization of external genitalia), partial AIS (mainly
female, mainly male or ambiguous external genitalia) and mild AIS (male
external genitalia and impaired pubertal virilization). AIS is the most
common disorder of sexual differentiation in individuals with 46XY
karyotype. Mutations in the androgen receptor gene are found in more
than 95% of individuals with complete AIS, while this occurs in 40% of
partial IAS cases. Children born with complete AIS have female external
genitalia, while those with partial IAS have atypical external
genitalia. The characteristic features of this disorder include a
female phenotype with normal breast development but absent or scanty
growth of pubic and axillary hair. The disorder also includes a vagina
of varying lengths along with the absence of the uterus, fallopian
tubes and ovaries. Testicular secretion of Müllerian inhibiting
substance suppresses development of the uterus, oviducts and upper
one-third of the vagina in utero. Gonads in the form of testes are
located at the internal inguinal ring, resides intraabdominal or can be
palpable in the labia majora in children with complete AIS. Complete
AIS is associated with amenorrhea and inguinal hernias in girls. The
diagnosis is established with karyotype analysis, imaging studies (US,
MRI) and a combination of hormonal dosages either at basal or after
gonadal stimulation. There is an increased risk of gonadal tumors in
patients with complete AIS. The invasive type II germ tumors
encountered are the seminoma if the gonad is testis, and dysgerminoma
if the gonad is considered an ovary. Seminoma is the most frequent
testicular tumor in complete AIS with an age at presentation of more
than 30 years. Currently, there are no clinically useful biomarkers
available to guide clinicians in predicting tumorous risk other than
direct gonadal histology and immunohistochemistry. If the gonads are
removed due to the risk of future malignancy, hormone replacement
therapy should be initiated and continued until the age of menopause.
There is discrepancy regarding the timing of gonadectomy in patients
with complete AIS. The consensus is to recommend delaying gonadectomy
until postpubertal status is reached to allow for spontaneous puberty
to develop through aromatization of testosterone into estrogen, since
there is a very low risk of malignancy before puberty. Gonadectomy
would necessitate initiation of hormone replacement therapy since
androgens are necessary for skeletal development. Therefore AIS
patients would require estrogen replacement to achieve and/or maintain
normal bone mass. Delaying gonadectomy until patients are of an age to
make their own medical decision remains safe, especially since the risk
of malignancy before puberty is very low. Ultrasound surveillance
should be utilized to screen patients for malignancy, should they
decide to retain their gonads into adulthood.
References:
1- Patel V, Casey RK, Gomez-Lobo V: Timing of Gonadectomy in Patients
with Complete Androgen Insensitivity Syndrome-Current Recommendations
and Future Directions. J Pediatr Adolesc Gynecol. 29(4):320-5, 2016
2- Chaudhry S, Tadokoro-Cuccaro R, Hannema SE, Acerini CL, Hughes IA:
Frequency of gonadal tumours in complete androgen insensitivity
syndrome (CAIS): A retrospective case-series analysis. J Pediatr Urol.
13(5):498.e1-498.e6, 2017
3- Weidler EM, Linnaus ME, Baratz AB, et al: A Management Protocol for
Gonad Preservation in Patients with Androgen Insensitivity Syndrome. J
Pediatr Adolesc Gynecol. 32(6):605-611, 2019
4- Weidler EM, Baratz A, Muscarella M, Hernandez SJ, van Leeuwen K: A
shared decision-making tool for individuals living with complete
androgen insensitivity syndrome. Semin Pediatr Surg. 28(5):150844, 2019
5- Lanciotti L, Cofini M, Leonardi A, Bertozzi M, Penta L, Esposito S:
Different Clinical Presentations and Management in Complete Androgen
Insensitivity Syndrome (CAIS). Int J Environ Res Public Health.
16(7):1268, 2019
6- Nemivant SM, van Leeuwen K, Weidler EM: Two cases of gonad retention
in adolescent patients with complete androgen insensitivity syndrome
(CAIS). J Pediatr Surg Case Rep. 52:101332, 2020
7- Kubo H, Kozan H, Kawai M: Ultrasonography for inguinal hernia led to
the diagnosis of complete androgen insensitivity syndrome. Pediatr Int.
63(1):122-123, 2021
Anorchia
Congenital anorchia, also known as testicular regression or
vanishing syndrome, is defined as the absence of one or both testes in
a 46,XY individual with a male phenotype. Anorchia occurs unilateral in
97% of cases accounting for 10% of cases in which the testis is absent
from the scrotum or inguinal canal. Testes are impalpable in 20% of
cryptorchidism cases, with unilateral anorchia as the cause in 35-60%
of them. Unilateral anorchia occurs in one of 5000 males. Bilateral or
true congenital anorchia is rare, occurs in one of each 180 cases of
cryptorchidism, or one in 20,000 male births. A few children with
anorchia present with ambiguous genitalia or microphallus. Anorchia is
a component of several malformation syndromes such as Cross syndrome,
OEIS syndrome, Saldino syndrome and sirenomelia. Phenotyping into male
external genitalia depends on anti-müllerian hormone (AMH) produce
by Sertoli cells and testosterone produced by Leydig cells. This means
that testes were present but disappeared in utero. The genetic cause of
anorchia is not known. Laparoscopy has suggested that some cases of
anorchia are the result of prenatal testicular vascular accidents
associated with torsion during in-utero testicular descent. Infants
with bilateral anorchia present with micropenis in almost 50% of cases.
Upon examination palpable testes are absent, while during laparoscopy
blind-ending spermatic cord and epididymides are usually present. In
children with bilateral anorchia serum testosterone concentration is
very low and does not increase in response to HCG stimulation. Serum
AMH concentrations are usually undetectable in patients with bilateral
congenital anorchia. Inhibin B is undetectable in most boys with
bilateral congenital anorchia. Undetectable levels of AMH and inhibin
B, along with elevated FSH and LH levels in a 46,XY karyotype is
sufficient evidence for diagnosis of congenital bilateral anorchia.
True bilateral anorchia must be differentiated from intra-abdominal
bilateral cryptorchidism. Diagnosis is based on a combination of
biochemical tests, karyotype, imaging studies and surgical/laparoscopic
exploration. Surgical/laparoscopic exploration and histologic findings
typically show nubbins of fibrous tissue devoid of any testicular
tissue attached to a blind-ending vas deferens. Histopathology
examination has confirmed the presence of germ cells in 0-16% of
excised testicular remnants. Germ cell tumors cannot develop from a
testis remnant that has no germ cell survival from the early embryonic
primordial germ cells. Hence tumor development is extremely rare in
remnants with germ cells. Nubbin excision should be performed if the
internal ring is open with normal vessels. Management of congenital
bilateral anorchia consists of testosterone replacement to stimulate
penile length and induce sexual development. Testicular prostheses can
be implanted in the scrotum for psychological and cosmetic reasons.
Unilateral anorchia does not require hormonal management.
References:
1- Brauner R, Neve M, Allali S, et al: Clinical, biological and genetic
analysis of anorchia in 26 boys. PLoS One. 6(8):e23292, 2011
2- Teo AQ, Khan AR, Williams MP, Carroll D, Hughes IA: Is surgical
exploration necessary in bilateral anorchia? J Pediatr Urol.
9(1):e78-81, 2013
3- Pirgon O, Dundar BN: Vanishing testes: a literature review. J Clin Res Pediatr Endocrinol. 4(3):116-20, 2012
4- Woodford E, Eliezer D, Deshpande A, Kumar R: Is excision of
testicular nubbin necessary in vanishing testis syndrome? J Pediatr
Surg. 53(12):2495-2497, 2018
5- Jespersen K, Ljubicic ML, Johannsen TH, et al: Distinguishing
between hidden testes and anorchia: the role of endocrine evaluation in
infancy and childhood. Eur J Endocrinol. 183(1):107-117, 2020
6- Shin J, Jeon GW: Comparison of diagnostic and treatment guidelines
for undescended testis. Clin Exp Pediatr. 63(11):415-421, 2020
Mixed Gonadal Dysgenesis
Mixed gonadal dysgenesis (MGD) is a very rare disorder of
sexual development characterized by gonadal asymmetry with an abnormal
dysgenetic testis on one side and a streak gonad on the contralateral
side. Phenotypic features of MGD is variable, and include normal males,
females with or without turner-like physical characteristic, and cases
of ambiguous genitalia. Most common MGD karyotype includes a
45,XO/46,XY mosaicism. Rare mosaic karyotype identified in MGD can
include 45,XO/47,XYY or 45XO/46XY/47XYY. The phenotypic abnormalities
are the result of incomplete inhibition of müllerian structures,
and incomplete masculinization of external genitalia. 95% of MGD
children have müllerian remnants and 75% of streaks gonads have an
ipsilateral fallopian tube. 90-95% of patients with a prenatal
diagnosis of 45,XO/46XY will be phenotypically normal male.
Clinically they present as children with ambiguous or abnormal
genitalia, or adults with gonadal failure or short stature. Other
associated problems in MGD include cardio renal malformations, gonadal
blastomas and germ cell tumors. Patients with bilateral streaks are
associated with the phenotype of a sexually infantile female, those
with a streak and intra-abdominal testis present with clitoromegaly in
a female, and those with one scrotal testis and an intraabdominal
streak are associated with frank sexual ambiguity, and bilateral
scrotal testis present as a male with short stature and gonadal
failure. All of these cases with MGD are infertile. Diagnosis should be
suspected with delay in puberty changes, short stature, webbed neck,
and coarctation of the aorta. Diagnosis is established with karyotype,
cytogenetic studies (FISH or PCR analysis), imaging studies (US, MRI)
and laparoscopy. During laparoscopy a biopsy of each gonad should be
ascertained before embarking in bilateral gonadectomy. In MGD, the
gonadal phenotype ranges from streak gonads through dysgenetic to
functioning testes. Congenital adrenal hyperplasia should be
rule-out clinically and biochemically. In patients with MGD the sex of
rearing decision is usually female. The term Y-chromosome gonadal
dysgenesis is used for both 46,XY and 45,XO/46,XY karyotype with MGD.
Early correct diagnosis of Y-chromosome gonadal dysgenesis has a higher
potential malignant risk. The risk of developing malignancy depends on
how much Y material is present. The specific location on the Y
chromosome that has been identified is the gonadoblastoma location
known as the GBY region. Gonadal tumor development is one of the most
important challenges in patients with MGD. The most common tumor
observed is gonadoblastoma, followed by invasive germ cell tumor.
Gonadectomy for the Y-chromosome gonadal dysgenesis should be
accomplished during the first decade of life since most tumors develop
during the second decade. Neoplastic transformation of a germ cell in
dysgenetic gonads, either gonadoblastoma or invasive germ cell tumor,
occurs in 20-30% of 46,XY MGD patients.The child to be raised as a
female will need clitoral recession and vaginoplasty in early infancy.
If it is to be raised as male, then various types of hypospadias repair
can be done, gonads can be replaced with prostheses, the prepenile
scrotum reconstructed and Müllerian structures removed.
References:
1- Chertin B, Koulikov D, Alberton J, Hadas-Halpern I, Reissman P,
Farkas A: The use of laparoscopy in intersex patients. Pediatr Surg
Int. 22(5):405-8, 2006
2- Flannigan RK, Chow V, Ma S, Yuzpe A: 45,X/46,XY mixed gonadal
dysgenesis: A case of successful sperm extraction. Can Urol Assoc J.
8(1-2):E108-10, 2014
3- Mizuno K, Kojima Y, Kurokawa S, Mizuno H, Kohri K, Hayashi Y:
Laparoscopic diagnosis and treatment of a phenotypic girl with mosaic
45,XO/46,X,idic(Y) mixed gonadal dysgenesis. J Pediatr Surg. 44: E1-E3,
2009
4- Berberoglu M,Siklar Z; Ankara University Dsd Ethic Committee: The
Evaluation of Cases with Y-Chromosome Gonadal Dysgenesis: Clinical
Experience over 18 Years. J Clin Res Pediatr Endocrinol. 10(1):30-37,
2018
5- Weidler EM, Pearson M, van Leeuwen K, Garvey E: Clinical management
in mixed gonadal dysgenesis with chromosomal mosaicism: Considerations
in newborns and adolescents. Semin Pediatr Surg. 28(5):150841, 2019
6- Saikia UK, Sarma D, Das DV, et al: A Case of Mixed Gonadal
Dysgensis: A Diagnostic Challenge. J Hum Reprod Sci. 12(2):169-172, 2019
7- Leng XF, Lei K, Li Y, et al: Gonadal dysgenesis in Turner syndrome
with Y-chromosome mosaicism: Two case reports. World J Clin Cases.
8(22):5737-5743, 2020
PSU Volume 57 NO 04 OCTOBER 2021
Parastomal Hernia
Parastomal hernia (PH) is an incisional hernia that occurs
within the surrounding of a stoma where abdominal content, typically
bowel or omentum, protrude between the skin and bowel stomal edge
surrounded by a hernial sac. Parastomal hernia is a common complication
of various type of stomas. It can progress asymptomatic, resulting in
an abdominal deformity, but it can lead to bowel incarceration and
strangulation needing urgent surgery. In infants and young children the
most common indications for performing a stoma include necrotizing
enterocolitis, Hirschsprung's disease and anorectal malformations. In
adolescent children the indication is intractable functional
constipation, intestinal pseudo obstruction and inflammatory bowel
disease. An ostomy prolapse in children is more common than parastomal
hernia in children. Prolapse is more common after loop than end
enterostomies. Patients with gastrointestinal motility disorders have a
higher complication rate and more severe complications in comparison to
the children without gastrointestinal motility disorders. In adults the
two most common conditions associated with stoma construction include
colorectal cancer and inflammatory bowel disease. It is believed that
30 to 50% of stoma will develop a parastomal hernia, and one-third of
these cases will need surgical correction. End (colon) ostomies have a
higher probability of developing a parastomal hernia than loop (ileum)
ostomies. Risk factors associated with developing a parastomal hernia
include age above 60 years, obesity, diabetes, tobacco consumption,
systemic and local infection, COPD, steroid therapy, inflammatory bowel
disease and cancer. The incidence of parastomal hernia as a recurrence
after corrective surgery is lower when using mesh for the repair.
Diagnosis of a parastomal hernia is by physical examination. In the
vast majority of cases the only clinical symptom is a deformity of the
abdominal wall around the stoma. Parastomal hernia can be overlooked in
obese patients. The use of ultrasound, CT-Scan or MRI increases the
diagnostic accuracy. Indications for surgical management of parastomal
hernia are limited to those with severe symptoms and complications of
bowel obstruction occurring in 30% of all hernia patients. Indications
for surgical management include incarceration, strangulation,
obstruction, parastomal fistula, perforation, ischemia, recurrent
symptoms of obstruction, difficulty maintaining collection device,
hernia-related pain, and problems with irrigation of the stoma. Several
methods utilized for corrective surgery of a parastomal hernia include
open transposition of the stoma, use of mesh reinforcement, or repair
using minimally invasive technique. Transposition is associated with a
lower risk of hernia recurrence when compared with local
reconstruction. When mesh is utilized, it can be placed superficially
(onlay technique), pre-peritoneally (sublay technique) or
intraperitoneally (inlay technique). Laparoscopic technique relies on
intraperitoneally implanted prosthetics. Reinforcement of the abdominal
wall with prosthetic material is the method of choice since it promises
good results and low incidence of complications and recurrences over
long periods of time.
References:
1- Nour S, Beck J, Stringer MD: Colostomy complications in infants and children. Ann R Coll Surg Engl. 78(6):526-30, 1996
2- Jayarajah U, Samarasekara AM, Samarasekera DN: A study of long-term
complications associated with enteral ostomy and their contributory
factors. BMC Res Notes. 9(1):500, 2016
3- Ohashi K, Koshinaga T, Uehara S, Furuya T, et al: Sutureless
enterostomy for extremely low birth weight infants. J Pediatr Surg. 52:
1873-1877, 2017
4- Youseff F, Arbash G, Puligandla PS, Baird RJ: Loop versus divided
colostomy for the management of anorectal malformations: a systematic
review and meta-analysis. J Pediatr Surg. 52: 783-790, 2017
5- Vriesman MH, Noor N, Koppen IJ, Di Lorenzo C, de Rong JR, Beninga
MA: Outcomes after enterostomies in children with and without motility
disorders: A description and comparison of postoperative complications.
J Pediatr Surg. 55: 2413-2418, 2020
6- Andersen RM, Klausen TW, Danielsen AK, et al: Incidence and risk
factors for parastomal bulging in patients with ileostomy or colostomy:
a register-based study using data from the Danish Stoma Database
Capital Region. Colorectal Dis. 20(4):331-340, 2018
Juvenile Granulosa Ovarian Tumor
Granulosa cell tumors of the ovary are rare benign ovarian
sex cord-stromal tumors. Granulosa cell tumors are divided into a
juvenile granulosa cell and adult granulosa cell variety tumor. Ovarian
tumors account for approximately 1% of all tumors in children and
adolescent. Juvenile granulosa cell tumor (JGCT) accounts for 67% of
sex cord-stromal tumor in the pediatric population and
approximately 5-12% of all ovarian neoplasms in children. Nearly half
of the patients are diagnosed in the first decade of life with a median
age of presentation of 7 years. The clinical significance of JGCT is
due to its estrogen secreting properties resulting in pseudo precocious
puberty without ovulation. More than 80% of patients present with
symptoms of precocious puberty including increased pubic hair, vaginal
bleeding, breast enlargement and advanced bone age. In older ages and
adolescents JGCT causes other manifestations such as hirsutism,
abnormal uterine bleeding and abdominal discomfort. There is a high
level of sex hormones and suppressed gonadotropin level in this
condition. JGCT secretes estradiol due to the presence of theca cells
that secrete androstenedione which is subsequently converted to
estradiol by the granulosa cells. Inhibin A & B which are
synthesized by the granulosa cells are also elevated supporting the
diagnosis. A pelvic mass is usually present. The triad of a palpable
adnexal mass, elevated serum estradiol and absent or decreased
gonadotropin is almost diagnostic of JGCT. JGCT are usually large
(averaging 12 cm) and in most cases limited to the ovary. Under
ultrasound granulosa tumors are solid and cystic or mainly solid with a
spongiform appearance with the solid portion being heterogeneous in
echogenicity. On MRI the solid component is typically isodense and
enhances. Fluid-wave levels within the cystic component represent areas
of hemorrhage. Granulosa cell tumors of the ovary rare calcify or
spread to the peritoneum, unlike epithelial neoplasm. JGCT are
typically unilateral and confined to the affected ovary at diagnosis.
Hence, most cases (>90%) are diagnosed with FIGO Stage 1 which
respond well to unilateral salpingo-oophorectomy. Surgery should be
performed in this age group with unilateral oophorectomy only for stage
1. There is no role for simple ovarian cystectomy. Staging should
include peritoneal cytology, exploratory laparotomy and unilateral
salpingo-oophorectomy. Bilateral ovarian involvement is uncommon in
stage 1 tumors and wedge biopsy is not recommended. Prognostic factors
include the size of the tumor, degree of nuclear atypia and mitotic
activity. Tumor rupture is not a negative prognostic factor. Serum
estradiol, CA-125 and inhibin B may be used for follow-up
postoperatively. Precocious puberty changes subside and physiologic
puberty occurs at the normal expected age in all cases after tumor
removal. Advance disease might need cytoreductive surgery followed by
combination aggressive chemotherapy. Lymph node involvement is a rare
phenomenon in sex-cord stromal tumors.
References:
1- Hashemipour M, Moaddab MH, Nazem M, Mahzouni P, Salek M: Granulosa
cell tumor in a six-year-old girl presented as precocious puberty. J
Res Med Sci. 15(4):240-2, 2010
2- Fleming NA, de Nanassy J, Lawrence S, Black AY: Juvenile granulosa
and theca cell tumor of the ovary as a rare cause of precocious
puberty: case report and review of literature. J Pediatr Adolesc
Gynecol. 23(4):e127-31, 2010
3- Powell JL, Kotwall CA, Shiro BC: Fertility-sparing surgery for
advanced juvenile granulosa cell tumor of the ovary. J Pediatr Adolesc
Gynecol. 27(4):e89-92, 2014
4- Wu H, Pangas SA, Eldin KW, et al: Juvenile Granulosa Cell Tumor of
the Ovary: A Clinicopathologic Study. J Pediatr Adolesc Gynecol.
30(1):138-143, 2017
5- Hansen R, Lewis A, Sullivan C, Hirsig L: Juvenile granulosa cell
tumor diagnosed in 6-month-old infant with precocious puberty. Radiol
Case Rep. 16(9):2609-2613, 2021
6- Parikshaa G, Ariba Z, Pranab D, et al: Juvenile granulosa cell tumor
of the ovary: A comprehensive clinicopathologic analysis of 15 cases.
Ann Diagn Pathol. 2021 Jun;52:151721. doi:
10.1016/j.anndiagpath.2021.151721. Epub 2021 Feb 10.
Duct of Luschka
Ducts of Luschka are subvesical accessory biliary ducts
located in the gallbladder fossa. They branch from the right hepatic or
common hepatic duct, are not accompanied by artery or vein as other
bile ducts draining liver segments (a portal triad is absent). Ducts of
Luschka are small, less than 1-2 mm in diameter, usually originating
from the lower aspect of the right hepatic lobe running along the
gallbladder fossa and liver parenchyma. They do not open into the
gallbladder. Drainage may be into intrahepatic or extrahepatic biliary
ducts. Ducts of Luschka should be differentiated from hepatocystic
ducts which are aberrant ducts that could drain a significant amount of
liver parenchyma into the gallbladder or cystic duct. The reported
prevalence of duct of Luschka is 4%. Injury to the ducts of Luschka
during laparoscopic or open cholecystectomy can cause postoperative
bile leak and peritonitis. Most injuries to the duct of Luschka occur
after ligation and division of the cystic artery and cystic duct while
dissecting the gallbladder from the liver fossa. These ducts can also
be injured during liver resection, liver transplantation and
interventional radiological procedures. In very rare occasions the
ducts of Luschka can be identified intraoperatively. Patients with bile
leaks have variable clinical course presenting with mild abdominal
pain, tenderness, fever or biliary peritonitis with sepsis. There is
mild elevation of serum bilirubin and alkaline phosphatase. Timing of
presentation of such leaks is usually within the first postoperative
week. The patient with duct of Luschka leaks will develop a fluid
collection (biloma) diagnosed by US or CT-Scan. A percutaneous drainage
is usually necessary to drain the bile leak. Performing a fistulogram
through the draining catheter will demonstrate a communication with the
biliary tree. Though a HIDA scan will demonstrate a bile leak, it
cannot give an anatomical impression of where the bile leak is coming.
MRCP can diagnose a leaking duct of Luschka. ERCP is the standard mode
of diagnosing a duct of Luschka leak. ERCP can also be therapeutic by
reducing intrabiliary pressure with sphincterotomy and endobiliary
stent placement. The management of a duct of Luschka leak depends on
the clinical condition of the patient. Asymptomatic patients with a low
output leak can be managed with simple drainage. Spontaneous resolution
of the leak may occur because accessory ducts do not drain a
significant portion of the liver. Should the leak produce a higher
output of bile, ERCP with sphincterotomy, stenting or nasobiliary tube
placement should be in order. Patients with severe symptoms and those
where the leak persists despite endoscopic treatment should be
reexplored and ligation of the leaking duct
performed.
References:
1- Spanos CP, Syrakos T: Bile leaks from the duct of Luschka
(subvesical duct): a review. Langenbecks Arch Surg. 391(5):441-7, 2006
2- Masoni L, Landi L, Maglio R: Intraoperative Treatment of Duct of
Luschka during Laparoscopic Cholecystectomy: A Case Report and Revision
of Literature. Case Rep Surg. 2018 Dec 17;2018:9813489. doi:
10.1155/2018/9813489. eCollection 2018.
3- Paramythiotis D, Moysidis M, Rafailidis V, et al: Ducts of Luschka
as a rare cause of postoperative biloma. MRCP findings. Radiol Case
Rep. 14(10):1237-1240, 2019
4- Spanos CP, Spanos MP: Subvesical bile duct and the importance of the
critical view of safety: Report of a case. Int J Surg Case Rep.
2019;60:13-15. doi: 10.1016/j.ijscr.2019.05.040. Epub 2019
May 28.
5- Handra-Luca A, Ben Romdhane HM, Hong SM: Luschka Ducts of the
Gallbladder in Adults: Case Series Report and Review of the Medical
Literature. Int J Surg Pathol. 28(5):482-489, 2020
6- Oulad Amar A, Kora C, Jabi R, Kamaoui I: The Duct of Luschka: An
Anatomical Variant of the Biliary Tree - Two Case Reports and a Review
of the Literature. Cureus. 2021 Apr 25;13(4):e14681. doi: 10.7759
PSU Volume 57 NO 05 NOVEMBER 2021
Anorectal Myectomy
In 1964 Lynn device a surgical procedure for short segment
Hirschsprung's disease (HD). This procedure was later utilized for
chronic idiopathic constipation not associated with Hirschsprung's
disease and for children who suffered from anal sphincteric achalasia
after a pull-through procedure. A rectal biopsy is warranted in cases
of chronic constipation to determine the presence or absence of
ganglion cells. The biopsy can be performed using a suction biopsy tool
in children less than three months of age, while a full-thickness
biopsy might be needed for children above six months of age. Thus will
determine if the child has aganglionosis or not. The anorectal myectomy
consists of an outpatient procedure. Dissection of the internal
sphincter from the mucosa and external sphincter is performed in the
intersphincteric plane. The dissection is extended proximally for five
to 7 centimeters. Excision of one centimeter wide strip of the internal
sphincter muscle is performed for the length of the dissection. Barium
enema suggests the presence of ultra-short segment HD but may not
indicate precisely the extent of the disease and in some children a
constricting segment may not be demonstrable. Confirmation will only
take place after a rectal biopsy is performed. If after anorectal
posterior myectomy for HD there is no relief of symptoms then the
child will need a major procedure for cure. Anal stricture and
incontinence have been reported as complications of myectomy. The
advantage of this operation is it's relatively simplicity and in
addition serves as both diagnostic and therapeutic. Internal anal
sphincter achalasia is a clinical condition with a presentation similar
to HD. The diagnostic criteria for anorectal sphincter achalasia are
based on anorectal manometry showing absence of the recto-inhibitory
reflex associated with a normal rectal biopsy. Anorectal sphincter
achalasia reflects the failure of relaxation of the internal sphincter.
The exact pathogenesis and pathophysiology of internal sphincter
achalasia is not fully understood. Patients present with constipation
and soiling with or without abdominal distension. Nutritional support,
laxatives and enemas are the first line of treatment of chronic
constipation associated with achalasia of the sphincter and
approximately 85% of cases could improve or cure by conservative
medical management. Due to an inadequate response to medical treatment
of constipation other children are selected for surgery.
Anorectal myectomy relieves more than 60% of patients with chronic
refractory constipation associated with internal sphincter achalasia.
In children with internal anal sphincter achalasia, posterior anorectal
myectomy of the internal sphincter is a more effective treatment option
when compared with intrasphincteric Botox injection. Anorectal myectomy
is an effective and safe procedure in patients suffering from
persistent chronic constipation in spite of medical treatment. Is also
the definitive treatment for children and adults with
ultrashort-segment Hirschsprung's disease.
References:
1- Redkar RG, Mishra PK, Thampi C, Mishra S: Role of rectal myomectomy
in refractory chronic constipation. Afr J Paediatr Surg. 9(3):202-5,
2012
2- Mousavi SA, Karami H, Rajabpoor AA: Intractable chronic constipation
in children: outcome after anorectal myectomy. Afr J Paediatr Surg.
11(2):147-9, 2014
3- Friedmacher F, Puri P: Comparison of posterior internal anal
sphincter myectomy and intrasphincteric botulinum toxin injection for
treatment of internal anal sphincter achalasia: a
meta-analysis. Pediatr Surg Int. 28(8):765-71, 2012
4- Doodnath R, Puri P: Long-term outcome of internal sphincter myectomy
in patients with internal anal sphincter achalasia. Pediatr Surg Int.
25(10):869-71, 2009
5- Ortega Escudero M, Gutierrez Duenas JM, Hernandez Diaz C, et al:
[Outcome of posterior anorectal myectomy for the treatment of
idiopathic chronic constipation]. Cir Pediatr. 28(4):193-195, 2015
6- Siminas S, Losty PD: Current Surgical Management of Pediatric
Idiopathic Constipation: A Systematic Review of Published Studies. Ann
Surg. 262(6):925-33, 2015
Vaping Lung Injury
Electronic cigarettes are alternative tobacco products that
deliver nicotine without the tobacco smoke. They are devices that
produce an aerosol by heating a liquid that contains a solvent
(vegetable glycerin, propylene glycol) on one of several flavoring with
or without nicotine. Each device structurally comprises four
components: a battery, a reservoir for the liquid, a vaporizing chamber
with a heating element and a mouthpiece for inhalation. Evaporation of
the liquid during heating followed by rapid cooling forms the aerosol
which after inhaled is term "vaped". This method of smoking is less
harmful as compared with cigarette use since it's not associated with
inhalation of combustible products of tobacco which is more
carcinogenic. While most e-cigarettes deliver nicotine and a flavoring
agent, many contain cannabis-based compounds used as a substitute for
traditional marijuana. Vaping use has increased significantly between
high school and middle school students. Vaping has been associated with
increase odds of myocardial infarction, thermal injury due to
explosions, seizures and psychosocial effects due to addiction. Lately
electronic cigarettes have been associated with significant lung injury
(EVALI = electronic vaping associated lung injury), with the majority
of cases in teenage and young adult males. Vaping generates harmful
carbonyl compounds such as formaldehyde, acrolein and acetaldehyde
implicated in the development of oxidative stress and release of
inflammatory mediators causing airway epithelial injury. Most
associated vaping lung injury involves THC, the active ingredient in
marijuana and vitamin E acetate which is used a diluent. Vitamin E
acetate when heated and aerosolized produce ketene, an extreme irritant
of airways further propagating inflammation. Common respiratory
symptoms found in this patients include shortness of breath, cough,
pleuritic chest pain and hemoptysis. Gastrointestinal symptoms of
nausea and abdominal pain, associated with fever and chills are
reported in the majority of cases. This inflammation can progress to
hypoxemic respiratory failure decreasing oxygen saturation. Mechanical
ventilation will be required in 15-30% of these patients. Chest films
and CT-Scans show bilateral hazy or consolidate opacity with lower lobe
or centrilobular ground glass nodule appearance. Pleural effusions are
rarely seen. Vaping associated lung injury(EVALI) is a diagnosis
of exclusion with the case definition being a history of vaping within
90 days of symptom onset, abnormal imaging and absence of an
alternative diagnosis such as infection. An infectious workup should be
performed in all cases. Bronchoscopy with bronchoalveolar lavage with
transbronchial biopsy should be performed to exclude other causes of
injury. Pathology of specimen shows organizing pneumonia, diffuse
alveolar damage, lipoid pneumonia, acute fibrinous pneumonitis or a
combination of these patterns. Cytology of alveolar lavage revels foamy
macrophages and pneumocyte vacuolization. Labs evaluation reveals
leukocytosis, elevated inflammatory markers and elevated lactate
dehydrogenase levels. Management consists of antibiotics, steroids,
high-flow oxygen therapy, mechanical ventilation and extracorporeal
membrane oxygenation. Prognosis is excellent in young patients with
most improving after a week of therapy. Due to the alarming number of
EVALI cases a public crisis has been
declared.
References:
1- Cherian SV, Kumar A, Estrada-Y-Martin RM: E-Cigarette or Vaping
Product-Associated Lung Injury: A Review. Am J Med. 133(6):657-663, 2020
2- Fedt A, Bhattarai S, Oelstrom MJ: Vaping-Associated Lung Injury: A
New Cause of Acute Respiratory Failure. J Adolesc Health.
66(6):754-757, 2020
3- Gilley M, Beno S: Vaping implications for children and youth. Curr Opin Pediatr. 32(3):343-348, 2020
4- Thakrar PD, Boyd KP, Swanson CP, Wideburg E, Kumbhar SS:
E-cigarette, or vaping, product use-associated lung injury in
adolescents: a review of imaging features. Pediatr Radiol.
50(3):338-344, 2020
5- Chadi N, Minato C, Stanwick R: Cannabis vaping: Understanding the
health risks of a rapidly emerging trend. Paediatr Child Health.
25(Suppl 1):S16-S20, 2020
6- Rao DR, Maple KL, Dettori A, et al: Clinical Features of
E-cigarette, or Vaping, Product Use-Associated Lung Injury in
Teenagers. Pediatrics. 2020 Jul;146(1):e20194104. doi:
10.1542/peds.2019-4104.
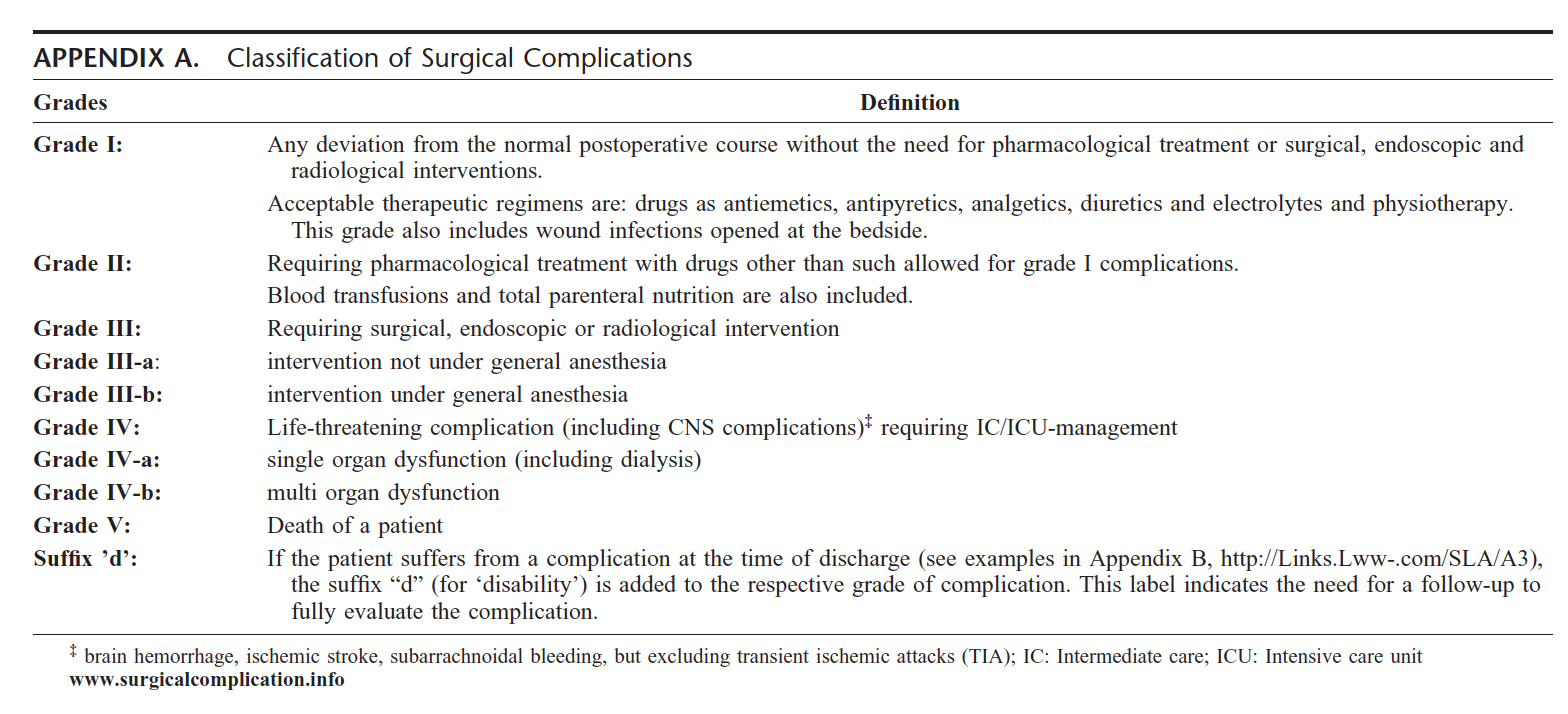
Clavien-Dindo Classification
The Clavien-Dindo classification (CDC) is a standardized
system for the registration of surgical complications. It was initially
based in three definitions of different outcomes after surgery: failure
to cure, sequelae and complications, completed by a system ranking
complications according to severity. Clavien focussed on grading the
severity of complications on the basis of the therapeutic consequence
required to manage the complication. The classification can be seen in
the Table below. There are five grades of evaluation of a surgical
complication. Grade 1 includes any deviation from the normal
postoperative course without the need for pharmacological treatment or
surgical, endoscopic or radiological intervention. Acceptable
therapeutic regimens include drugs as antiemetics, antipyretics,
analgesics, diuretics and electrolytes. This grade includes wound
infections opened bedside. Grade II includes requiring pharmacologic
treatment with drugs other than those allowed for Grade 1
complications. Grade II complications are those that result in
deviations from the normal postoperative course including unplanned or
additional clinic or office visits that can be managed as outpatients
without additional invasive, radiographic or surgical procedures. This
includes wound infections, transient nerve injury, deep-vein thrombosis
necessitating anticoagulation. Grade III requires surgical, endoscopic
or radiological intervention. Grade III is subdivided further into
III-A if intervention does not require general anesthesia, and Grade
III-B if the intervention requires general anesthesia. Grade IV is a
life threatening complication, including CNS complication or requiring
intensive care management. Grade IV is subdivided into Grade IV-A if it
involves single organ dysfunction (including dialysis) and Grade IV-B
if it includes multiorgan dysfunction. Grade V is death of a patient. A
suffix "d" is added to each grade if the patient suffers a complication
at the time of discharge. The label indicates the need for follow-up to
fully evaluate the complication. Clavien-Dindo Grade II is the most
represented complication overall accounting for almost 20% of all
patients. The CDC can be applied to patients who have undergone
elective and emergency surgery during the first 30 postoperative days.
Complication rates during emergency cases are higher than in elective
procedures. Complications are higher in neonates than in other
pediatric group. The advantage of this system is that all possible
adverse events are included. The Clavien-Dindo grading system while
widely used in general, transplantation and orthopedic surgery, it has
been sporadically used in pediatric surgery to identify complications
related to jejunal feeding, after repair of congenital duodenal
obstruction, after Nuss procedure, after ileostomy and colostomy
procedures, and after transanal endorectal pull-through for
Hirschsprung's disease. The most recurring complication in pediatrics
using the CDC is wound infection and post-appendectomy fluid
collection/abscess. A high complication rate after enterostomy
formation in children with motility disorders was identified using the
CDC. Virtually all current general surgical publications with morbidity
as an outcome measure use the Clavien-Dindo
classification.
References:
1- Clavien PA, Barjun J, de Oliveira ML, et al: The Clavien-Dindo
Classification of Surgical Complications. Five-Year Experience. Ann
Surg. 250: 187-196, 2009
2- Dodwell ER, Pathy R, Widmann RF, et al: Reliability of the Modified
Clavien-Dindo-Sink Complication Classification System in Pediatric
Orthopaedic Surgery. JB JS Open Access. 3(4):e0020, 2018
3- Hoff N, Wester T, Granstrom AL: Classification of short-term
complications after transanal endorectal pullthrough for Hirschsprung's
disease using the Clavien-Dindo-grading system. Pediatr Surg Int.
35(11):1239-1243, 2019
4- Thompson H, Jones C, Pardy C, Kufeji D, Nichols E, Murphy F,
Davenport M: Application of the Clavien-Dindo classification to a
pediatric surgical network. J Pediatr Surg. 55(2):312-315, 2020
5- Pio L, Rosati U, Avanzini S, et al: Complications of Minimally
Invasive Surgery in Children: A Prospective Morbidity and Mortality
Analysis Using the Clavien-Dindo Classification. Surg Laparosc Endosc
Percutan Tech. 27(3):170-174, 2017
6- Vriesman MH, Noor N, Koppen IJ, Di Lorenzo C, de Jong JR, Benninga
MA: Outcomes after enterostomies in children with and without motility
disorders: A description and comparison of postoperative complications.
J Pediatr Surg. 55(11):2413-2418, 2020
PSU Volume 57 NO 06 DECEMBER 2021
Acute Traumatic Coagulopathy
Trauma causes over 4 million deaths per year in the USA.
Most potentially preventable deaths are due to bleeding. Disruption of
the hemostatic equilibrium occurs at the moment of traumatic impact in
children and adults. Tissue injury and blood loss during trauma causes
an endogenous acute coagulopathy referred to as acute traumatic
coagulopathy (ATC). Traumatic injury generates dysfunction of the
coagulation, anticoagulation and fibrinolysis system, featuring a
hypocoagulant state with prolongation of the prothrombin time (PT)
and/or activated partial thromboplastin time (aPTT). The PT and INR
have been suggested as the more sensitive test to the multiple
coagulation factor deficiencies associated to ATC. ATC develops rapidly
and has been identified within minutes of injury. Severe tissue trauma
and systemic hypoperfusion are prerequisites for development of ATC.
The worst coagulopathy is seen in patients with injury severity scores
above 35 and base deficits less than 12 mEq/L. Others mediators such as
hypothermia, acidosis, and hemodilution develop later after injury due
to hemorrhage, hypoperfusion and exposure and resuscitation with
hypocoagulable products. Presence of ATC during hospital admission is
independently associated with fourfold higher mortality and
significantly greater transfusion requirements. The overall length of
mechanical ventilation, ICU and hospital stay is longer in injured
patients with acute traumatic coagulopathy versus those with normal
hemostasis on admission. Patients presenting with ATC have a mortality
approaching 50%. An INR greater than 1.3 on admission is the most
predictive of risk of death over other characteristics labs. The higher
the INR the higher the risk of mortality. Endogenous systemic
anticoagulation and fibrinolysis have emerged as probable mediator of
ATC. Coagulation is acutely impaired after injury, starting with
fibrinogen concentration declining rapidly. Systemic anticoagulation
via activation of protein C is the most important functional mediator
of ATC. Fibrinolysis is also a functional component of ATC. Injury and
hemorrhagic shock cause primary platelet impairment. The vascular
endothelium is an active participant in the pathophysiology of ATC as
it capture thrombin and accelerates protein C activation 1000-fold. ATC
is not a consumptive coagulopathy, since it's characterized by
dysfibrinogenemia, systemic anticoagulation, impaired platelets
function and hyperfibrinolysis. The most consumed coagulation factors
following injury are fibrinogen and factor V. Reproducing whole blood
by transfusing injured patients with a balanced ratio of packed blood
red cells, fresh frozen plasma and platelets while minimizing
crystalloid resuscitation is associated with a reduced mortality. High
doses of fresh frozen plasma (10-20 ml/kg) are recommended to control
the severe traumatic bleeding as soon as possible. FFP and PRBC at a
predetermined ratio of 1:2 is recommended. Platelet transfusions
are recommended to maintain a goal above 50K/L in polytrauma, and >
100K/L in central nervous system injury. Correction of
hyperfibrinolysis using tranexamic acid is the final component to
effective damage control
resuscitation.
References:
1- Frith D, Davenport R, Brohi K: Acute traumatic coagulopathy. Curr Opin Anaesthesiol. 25(2):229-34, 2012
2- Cap A, Hunt B: Acute traumatic coagulopathy. Curr Opin Crit Care. 20(6):638-45, 2014
3- Cohen MJ: Acute traumatic coagulopathy: clinical characterization
and mechanistic investigation. Thromb Res. 133 Suppl 1:S25-7, 2014
4- Duan K, Yu W, Li N: The Pathophysiology and Management of Acute
Traumatic Coagulopathy. Clin Appl Thromb Hemost. 21(7):645-52, 2015
5- Simmons JW, Powell MF: Acute traumatic coagulopathy: pathophysiology
and resuscitation. Br J Anaesth. 117(suppl 3):iii31-iii43, 2016
6- Maegele M: The Diagnosis and Treatment of Acute Traumatic Bleeding and Coagulopathy. Dtsch Arztebl Int. 116(47):799-806, 2019
PICC Lines
Vascular access is a very important aspect of care for
children and adults. Peripheral inserted central venous catheters
(PICC) lines are required by almost one-third of neonates and children
admitted to intensive care units. Indications for PICC lines include
intravenous access, long-term antibiotics therapy, infusion of blood
products and total parenteral nutrition. Using ultrasound guidance,
PICC lines are easy and safe to insert due to placement in a peripheral
vein in the upper limb (cephalic or basilic vein) with the tip at a
central location in the superior or inferior vena cava
allowing high osmolality solutions to be delivered. Risk of hemothorax
and pneumothorax associated to central line placement is also avoided.
The use of the axillary vein for PICC line insertion in premature
neonates can significantly reduce the frequency of complications.
Infants with abdominal surgical pathology who have PICC lines placed in
the lower limb are at greater risk for major complications related to
venous thromboembolism. PICC lines are inserted ultrasound-guided
either using the modified Seldinger technique or the direct
sheathed-needle puncture technique. Both have similar complication
rates. The tip of the PICC lines is confirmed with a standard chest
film. The most common complications of PICC lines include in order of
frequency local inflammation at the site of insertion (redness and
swelling), infection with sepsis, thromboembolism and mechanical. An
infection occurs when there is a positive peripheral or central blood
culture or a positive catheter tip culture after removal in the
presence of clinical signs of catheter-related sepsis. The surgical
neonate has a PICC infection rate of 10-25% comparable to the infection
rate of Broviac catheters. Coagulase-negative staphylococcus is the
most common organism isolated from positive cultures in PICC lines.
Attempted catheter sterilization with antibiotics can lead to
complicated bacteremia. Complicated bacteremia is defined as the
presence of end-organ damage, multiple positive blood cultures or
death. End-organ damage is defined as presence of osteomyelitis, vital
organ abscess, positive echocardiogram with vegetation, or a positive
lumbar puncture. Lack of improvement of inflammatory markers or two
positive blood cultures in spite antibiotics therapy for sepsis needs
line removal. Recommendations to reduce the incidence of catheter
associated bloodstream infection (CABSI) include cleaning hands before
placement, wearing full barrier precautions during insertion, using
chlorhexidine to clean the skin, using prepackaged insertion bundles
and assessing the daily need for the line. There is also a decrease in
CABSI when lines are placed in the operating room. Withdrawing blood
from catheters less than 3 Fr.increase the occlusion rate of PICC
lines. The use of central lines is the most common cause for thrombosis
in neonates and infants preterm babies. Catheter-related venous
thromboembolism can be asymptomatic or can result in complications such
as deep vein thrombosis, portal vein thrombosis, Budd-Chiari, superior
vena cava syndrome, intracardiac thrombosis or pulmonary embolism.
References:
1- Njere I, Islam S, Parish D, Kuna J, Keshtgar AS: Outcome of
peripherally inserted central venous catheters in surgical and medical
neonates. J Pediatr Surg. 46(5):946-50, 2011
2- Panagiotounakou P, Antonogeorgos G, Gounari E, et al: Peripherally
inserted central venous catheters: frequency of complications in
premature newborn depends on the insertion site. J Perinatol.
34(6):461-3, 2014
3- Kisa P, Ting J, Callejas A, Osiovich H, Butterworth SA: Major
thrombotic complications with lower limb PICCs in surgical
neonates. J Pediatr Surg. 50(5):786-9, 2015
4- Freeman JJ, Gadepalli SK, Siddiqui SM, Jarboe MD, Hirschl RB:
Improving central line infection rates in the neonatal intensive care
unit: Effect of hospital location, site of insertion, and
implementation of catheter-associated bloodstream infection
protocols. J Pediatr Surg. 50(5):860-3, 2015
5- Dasgupta N, Patel MN, Racadio JM, Johnson ND, Lungren MP: Comparison
of complications between pediatric peripherally inserted central
catheter placement techniques. Pediatr Radiol. 46(10):1439-43, 2016
6- Rainey SC, Deshpande G, Boehm H, Camp K, Fehr A, Horack K, Hanson K:
Development of a Pediatric PICC Team Under an Existing Sedation
Service: A 5-Year Experience. Clin Med Insights Pediatr. 13: 1-5, 2019
7- Furlong-Dillard J, Aljabari S, Hirshberg E: Diagnostic accuracy
among trainees to safely confirm peripherally inserted central catheter
(PICC) placement using bedside ultrasound. Br J Nurs. 29(19):S20-S28,
2020
Congenital Thrombophilia
Thrombophilia is defined as an increased risk of developing
hypercoagulability and venous thrombosis. Thrombophilia may be
congenital or acquired. Although thrombosis may occur in either or both
venous and arterial vessels, the term thrombophilia is usually utilized
for venous thromboembolism (VTE). Main causes of congenital
thrombophilia are classified as loss of function such as deficiency of
antithrombin, protein C and protein S, or a gain of function such as
activated protein C resistance due to factor V Leiden mutation,
hyperprothrombinemia due to presence of the prothrombin mutation, or
dysfibrinogenemia due to impairment of the relevant metabolic pathway.
Acquired risk factors for developing thrombophilia include
antiphospholipid antibodies, detected as lupus anticoagulants and/or
anticardiolipin antibodies and/or anti-B-2-glycoprotein-I antibodies.
Laboratory testing for thrombophilia should be undertaken in any young
patients who experience an unprovoked thrombotic event and those with
recurrences. The identification of risk factors may permit genetic
counseling, modification of the patient lifestyle to avoid future risk
and the ability to identify relatives at risk. Thrombophilia testing
does not substantially help to predict or reduce the incidence of
thrombosis recurrence. The intensity and duration of anticoagulation
treatment for thrombophilia after a thrombotic event should not be
altered irrespective of the presence or absence of most thrombophilia
risk factors. It is recommended that thrombophilia testing be offered
to patients with a first VTE < than 50 years of age, recurrent VTE,
VTE at any age with a strong family history of thrombotic disease and
VTE occurring in unusual sites such as hepatic, mesenteric, portal and
cerebral veins. It is also recommended testing be offered to women
suffering VTE in association with pregnancy, the immediate postpartum
period, or oral contraceptive use. Most pediatric VTE are associated
with indwelling catheters and/or underlying medical conditions,
including congenital heart disease, inflammation, immobilization,
thrombophilia or malignancy. Portal vein thrombosis is associated with
umbilical venous catheter placement in neonates and is likely very
common. Other thrombotic conditions in neonates include purpura
fulminans, renal vein thrombosis and cerebral sinovenous thrombosis.
Deep vein thrombosis is the most common presentation of VTE in children
and indwelling catheterization is the most common trigger factor.
Congenital heart disease predisposes to both venous and arterial
thrombosis, especially in shunting lesions and those requiring
catheterization, surgery and/or ECMO. Arterial thrombosis is uncommon
in pediatrics and is nearly always associated with arterial trauma or
catheterization. In cases of thrombophilia the goal of therapy should
be rapid restoration of blood flow to reduce late effects. For
treatment of VTE most children receive either six weeks to six months
of therapeutic anticoagulation. Tissue plasminogen activator (tPA) is
utilized for thrombolysis of venous or arterial thrombi that are life,
limb or organs threatening. Antiplatelet agents are used in pediatrics
to prevent arterial thrombosis or thrombosis associated with congenital
heart disease. Patients with cancer with a central venous catheter and
factor V Leiden mutation have a higher risk of developing
catheter-related thrombosis. Severe thrombophilia might increase the
risk of thrombosis in Covid-19 patients.
References:
1- Tripodi A: A review of the clinical and diagnostic utility of
laboratory tests for the detection of congenital thrombophilia. Semin
Thromb Hemost. 31(1):25-32, 2005
2- Favaloro EJ, McDonald D, Lippi G: Laboratory investigation of
thrombophilia: the good, the bad, and the ugly. Semin Thromb Hemost.
35(7):695-710, 2009
3- Trenor CC 3rd: Thrombosis and thrombophilia: principles for
pediatric patients. Blood Coagul Fibrinolysis. 21 Suppl 1:S11-5, 2010
4- Boersma RS, Hamulyak K, Cate HT, Schouten HC: Congenital
thrombophilia and central venous catheter-related thrombosis in
patients with cancer. Clin Appl Thromb Hemost. 16(6):643-9, 2010
5- Alameddine R, Nassabein R, Le Gal G, et al: Diagnosis and management
of congenital thrombophilia in the era of direct oral anticoagulants.
Thromb Res. 185:72-77, 2020
6- de la Morena-Barrio ME, Bravo-Perez C, de la Morena-Barrio B, et al:
A pilot study on the impact of congenital thrombophilia in COVID-19.
Eur J Clin Invest. 51(5):e13546, 2021