PSU Volume 58 NO 01 JANUARY 2022
Completion Thyroidectomy
Thyroid nodules in children are managed based exclusively on
the results of fine-needle aspiration (FNA) biopsy. Benign FNA results
can be observed depending on the size, complexity and symptomatology of
the thyroid nodule. FNA results with positive pathology for papillary
carcinoma is best managed with total thyroidectomy with central lymph
node dissection. With results of FNA reported as indeterminate,
suspicious, insufficient or a follicular neoplasm, removal of the
affected lobe (hemithyroidectomy) might be performed. During removal of
the affected lobe, the contralateral neck and remaining lobe should not
be explored or violated. In cases in which a hemithyroidectomy is
performed and the final pathology is reported as a well-differentiated
papillary or follicular thyroid cancer, a completion thyroidectomy is
performed. Completion thyroidectomy reduces locoregional recurrence,
distant metastasis as well as low-risk carcinoma. The overall need for
completion thyroidectomy in the era of FNA is generally less than 5-10%
after lobectomy. The current major indications for completion
thyroidectomy are: gross extrathyroidal extension on the ipsilateral
side, gross residual disease on the esophagus, recurrent laryngeal
nerve or the tracheal wall, major vascular or capsular invasion and
poorly differentiated carcinoma or aggressive Hurthle cell carcinoma.
Whenever a completion thyroidectomy is to be performed, the surgeon
should study the gland previously removed to determine the status of
the parathyroid glands that might be removed with the specimen. The
rates of both temporary and permanent hypoparathyroidism are
considerably higher in patients in whom the parathyroid glands were
removed with the specimen. At the tine of completion thyroidectomy when
there is already a parathyroid gland in the initial hemithyroidectomy
specimen, the surgeon must make every effort to identify and preserve
both parathyroid and in the event of a suspected devascularization, the
gland should be autotransplanted. Hemithyroidectomy followed by
completion thyroidectomy does not appear to be associated with an
increase operative risk of hypocalcemia or recurrent laryngeal nerve
injury. The lower rate of temporary hypoparathyroidism and hypocalcemia
seen in the completion thyroidectomy group can be attributed to the
fact that the interval between operations allowed for recovery of any
reversible injury caused at the initial hemithyroidectomy.
Devascularized parathyroid glands require approximately four weeks to
return to full function. Compared with total thyroidectomy, completion
thyroidectomy has been associated with similar rates of recurrent
laryngeal nerve injury and lower rates of hypoparathyroidism. After
performing completion thyroidectomy, serum thyroglobulin levels tend to
be an adequate prognostic follow-up marker. Beside the effectiveness of
radioactive iodine for ablation of the remaining normal tissue or
residual microscopic disease is enhanced after completion
thyroidectomy. Recurrent laryngeal and superior laryngeal nerve
monitoring during completion thyroidectomy is associated with a
decrease risk of injury. Completion thyroidectomy is a safe procedure
with acceptable morbidity in the hand of experience surgeons.
References:
1- Rafferty MA, Goldstein DP, Rotstein L, et al: Completion
thyroidectomy versus total thyroidectomy: is there a difference in
complication rates? An analysis of 350 patients. J Am Coll Surg.
205(4):602-7, 2007
2- Gulcelik MA, Dogan L, Guven EH, Akgul GG, Gulcelik NE: Completion
Thyroidectomy: Safer than Thought. Oncol Res Treat. 41(6):386-390, 2018
3- Nicholson KJ, Teng CY, McCoy KL, Carty SE, Yip L: Completion
thyroidectomy: A risky undertaking? Am J Surg. 218(4):695-699, 2019
4- Sena G, Gallo G, Innaro N, et al: Total thyroidectomy vs completion
thyroidectomy for thyroid nodules with indeterminate
cytology/follicular proliferation: a single-centre experience. BMC
Surg. 19(1):87, 2019
5- Shaha AR, Michael Tuttle R: Completion thyroidectomy-indications and complications. Eur J Surg Oncol. 45(7):1129-1131, 2019
6- Shaha AR, Patel KN, Michael Tuttle R: Completion thyroidectomy-Have
we made appropriate decisions? J Surg Oncol. 123(1):37-38, 2021
Hematocolpos after Cloacal Repair
In females the most common anorectal malformation is an
imperforate anus with a rectovestibular fistula, followed by
rectoperineal fistula and the cloacal anomaly. Cloacal repair entails
reconstruction of the urethra, vagina and rectum which end in a common
channel. The aim of vaginal reconstruction is to provide a cosmetically
satisfactory introitus, a conduit for normal menstruation and pain-free
penetrative intercourse. There is a strong association of gynecologic
anomalies (60%) associated with cloaca. A high rate of menstrual
obstruction (40%) at puberty is well described. In less complex
malformations such as rectovestibular and rectoperineal fistula a
vaginal septum is the most common associated finding and can be managed
most effectively during the initial repair of the rectum without trauma
to the hymen or introitus. Vaginoscopy allows evaluation of the vaginal
anatomy during infancy and puberty by documenting of vaginal
duplication with 2 hemivaginas, and a septum, documentation of the
septal length and total vaginal length. During vaginoscopy, perhaps at
colostomy closure or creation of an appendicostomy, documentation of
the presence of mucus at the outer part of the cervix can be evaluated.
Also cannulation of the distal fallopian tube with instillation of
saline can be performed to visualize the egress of saline from the
vagina and confirm patency of the Müllerian system. Hematocolpus
is a medical condition of blood retained in the proximal vagina due to
an outflow tract obstruction or blockage of menstrual flow. The most
common cause of hematocolpus in children without anorectal malformation
is an imperforate hymen. In cases of repaired cloaca a longitudinal or
transverse vaginal septum, congenital or acquired vaginal atresia or
severe vaginal stricture, uterus didelphys and septate uterus can cause
hematocolpus. Ovarian function is normal in girls with repaired cloaca
so pubertal and breast development occurs as expected. Thelarche
(breast development) occurs between 9-10 years of age with menstruation
occurring at 12-12.5 years of age. Confirmation of the patency of the
reproductive tract before menarche is important to avoid obstruction,
pain, and risk to reproductive organs with infertility. It is during
this time that repaired cloaca can be studied further to determine if
there could exist the possibility of menstrual flow tract obstruction
and development of hematometrocolpos by performing serial ultrasound
studies. Almost 20% of children born with cloaca develop amenorrhea due
to absence of or underdeveloped Müllerian structures. US
surveillance of the reproductive structures should begin 6-9 months
after thelarche and continue every 6 months through menarche. If an
obstruction to menstrual flow is detected by visualization of a
thickened endometrium with hematocolpos, medical intervention should be
initiated immediately to minimize adverse sequelae. Hormonal
suppression of menses and endometrial stimulation should be started if
there is menstrual flow obstruction to prevent continued accumulation
of blood. Surgery is usually necessary to establish an adequate outflow
tract either by resection of a vaginal septum, introitoplasty,
posterior vaginoplasty or vaginal replacement with bowel in cases of
absent vagina. Other gynecologic concern for pubertal females include
the development of adnexal cysts, hydrosalpinges, endometriosis and
chronic pelvic pain. Women who had a history of a cloacal anomaly
should be delivered by cesarean section.
References:
1- Breech L: Gynecologic concerns in patients with anorectal malformations. Seminars Pediatr Surg. 19: 139-145, 2010
2- Versteegh HP, Sutcliffe JR, Sloots EJ, Wijnen RMH, Blaauw Id:
Postoperative complications after reconstructive surgery for cloacal
malformations: a systematic review. Tech Coloproctol 19: 201-207, 2015
3- Breech L: Gynecologic concerns in patients with cloacal anomaly. Seminars Pediatr Surg. 25: 90-95, 2016
4- Vilanova-Sanchez A, Reck CA, McCracken KA, et al: Gynecologic
anatomic abnormalities following anorectal malformations repair. J
Pediatr Surg. 53: 698-703, 2018
5- Vilanova-Sanchez A, McCracken K, Halleran DR, et al: Obstetrical
Outcomes in Adult Patients Born with Complex Anorectal Malformations
and Cloacal Anomalies: A Literature Review. J Pediatr Adolesc Gynecol.
32: 7-14, 2019
6- Fanjul M, Lancharro A, Molina E, Cerda J: Gynecological anomalies in
patients with anorectal malformations. Pediatr Surg Int. 35: 967-970,
2019
Apple Peel Atresia
Jejunoileal atresias are classified as type I characterized
by a transluminal septum; type II involves a fibrous cord connecting
two blind ending pouches; type IIIA has a V-shaped mesenteric defect;
and type IV exhibits multiple atetric segments. The term Apple peel
atresia (or Type IIIB intestinal atresia), occurring in less than 10%
of all jejunoileal atresias, refers to the development of a high
jejunal atresia with discontinuity of the small bowel and a wide gap in
the mesentery. The distal segment of jejuno-ileum is shortened and
assumes a helical configuration around a retrograde perfusing vessel.
The appearance is similar to a Christmas tree, hence the synonym
Christmas tree deformity. An intrauterine vascular accident after
emergence of the middle colic artery to the affected proximal bowel
during late gestation has been accepted as the cause of apple-peel
atresia presenting with a wide spectrum of occlusions of one or more
branches of the superior mesenteric artery. The bowel distal to the
atresia is precariously supplied in a retrograde fashion by anastomotic
arcades from the ileocolic, right colic or inferior mesenteric artery.
Most of these children have less than half of the normal length of the
small bowel and a physiological short bowel. Apple peel atresia is
usually reported as an isolated malformation, but has also been related
to malrotation, situs inversus and polysplenia. Not all distal
apple-peel atresias are associated with a proximal bowel atresia since
a few scattered reports of a mesenteric defect associated with a
marginal artery may cause the coiling defect of the apple peel as the
bowel outgrows it blood supply causing problems of ischemia later in
life associated with mesenteric internal hernias. Also, not all
apple-peel atresia are from the proximal jejunum, since very few have
been described arising after a proximal duodenal atresia related to the
second portion of the duodenum with absence of the third and fourth
portions of duodenum and superior mesenteric artery. Infants born with
high jejunal atresia have considerable dilatation of the proximal
bowel, while the distal segment is small and collapsed. Anastomosis
between two discrepant bowel sizes can cause functional bowel
obstruction. The resulting peristalsis is incapable of producing an
adequate upstream pressure gradient. As alternative, an antimesenteric
reduction-tapering proximal jejunoplasty can be used to perform
anastomosis reducing the caliber of the proximal bowel to fit an almost
end to end anastomosis with the distal microbowel. Serial transverse
enteroplasty of the dilated proximal jejunal atresia can also be
performed to reduce the caliber while lengthening it appropriately
without significant loss of absorptive area. Apple-peel atresias
frequently have a high incidence of prematurity, short gut, multiple
atresias and associated anomalies which constitute potential prognostic
factors. Antenatal US may suggest the diagnosis of jejunoileal atresia
with the presence of dilated fluid-filled loops of bowel and
polyhydramnios. Surgery is the preferred mode of treatment for
jejunoileal atresias and the goal of treatment is to establish bowel
continuity while preserving as much bowel length as possible.
Postoperative complications associated with apple-peel atresia include
anastomotic dysfunction (most common), sepsis (from leak or TPN), short
bowel syndrome, necrotizing bowel, stenosis and even death. Survival is
above 90%.
References:
1- Llore N, Tomita S: Apple peel deformity of the small bowel without atresia in a congenital
mesenteric defect. J Pediatr Surg. 48(1):e9-11, 2013
2- Onofre LS, Maranhao RF, Martins EC, Fachin CG, Martins JL:
Apple-peel intestinal atresia: enteroplasty for intestinal lengthening
and primary anastomosis. J Pediatr Surg. 48(6):E5-7, 2013
3- Sasa RV, Ranko L, Snezana C, Lidija B, Djordje S: Duodenal atresia
with apple-peel configuration of the ileum and absent superior
mesenteric artery. BMC Pediatr. 16(1):150, 2016
4- Dao DT, Demehri FR, Barnewolt CE, Buchmiller TL: A new variant of type III jejunoileal atresia.
J Pediatr Surg. 54(6):1257-1260, 2019
5- Zhu H, Gao R, Alganabi M, et al: Long-term surgical outcomes of apple-peel atresia. J Pediatr Surg. 54(12):2503-2508, 2019
6- Mangray H, Ghimenton F, Aldous C: Jejuno-ileal atresia: its
characteristics and peculiarities concerning apple peel atresia,
focused on its treatment and outcomes as experienced in one of the
leading South African academic centres. Pediatr Surg Int. 36(2):201-207, 2020
7- Zvizdic Z, Popovic N, Milisic E, Mesic A, Vranic S: Apple-peel
jejunal atresia associated with multiple ileal atresias in a preterm
newborn: A rare congenital anomaly. J Paediatr Child Health.
56(11):1814-1816, 2020
PSU Volume 58 No 02 FEBRUARY 2022
Pectus Arcuatum
Anterior chest wall deformities are not rare. They consist
of pectus excavatum as the most frequent form of chest deformities, and
by protrusion deformities such as pectus arcuatum and carinatum. Pectus
arcuatum, also known as pouter pigeon chest, Currarino-Silverman
syndrome, chondro-manubrial deformity or type two pectus carinatum, is
a rare and complex congenital chest wall deformity whose main feature
is protrusion and early ossification of the sternal angle of Lewis
associated with bilateral deformity of the 2nd to 4th cartilages. It
may also be associated with a depressed lower sternum. It also
involves a wavelike deformity, a mixed form of excavatum and carinatum
features, either along a longitudinal or along a transverse axis. The
visual appearance of pectus arcuatum is formed by the costal cartilage
protrusion. In most cases pectus arcuatum is a cosmetic defect. Though
the diagnosis is established by physical exam, chest films and CT-Scan
with 3-D reconstruction are needed if repair of the sternal defect is
warranted. Imaging using thoracic CT scans and customized aided design
virtual simulation allows the surgeon to predetermine the specific
cutting angle for each patient and therefore design a cutting template
tailored to the individual deformity. The surgical repair of this
rare deformity requires a modified open Ravitch technique. Patients are
usually young adults without comorbidities and no special preparation
is needed. The basic steps in the surgical correction described by
Ravitch consist of bilateral parasternal and subperichondrial resection
of the deformed costal cartilages, detachment of the xiphoid process,
transverse wedge osteotomy at the upper edge of the sternal depression,
and bending of the sternum to straighten its course, securing the
corrected position of the sternum. Satisfactory overall results occur
in 98% of patients. The Ravitch technique has a risk of growth
limitation to the thoracic cage due to a wide resection of the rib
cartilages, the reason that the repair is not undertaken until the
child has acquired a rigid skeletal structure later in life. Pectus
arcuatum can be successfully corrected by Ravitch-type of
chondrosternoplasty. Due to necessity to resect cartilages, late
puberty or adulthood is preferred, since by that age the growth of ribs
have finished. Repair of pectus deformity in children that might need
future cardiac surgery has revealed that concomitant surgery is
contraindicated before adolescence because pectus deformities may
spontaneously disappear or recur after early sternal surgery.
Congenital heart defects are reported occasionally as well as
simultaneous Poland syndrome. Concomitant surgery of cardiac defects
and pectus deformity is a reliable strategy in adolescent and adults
offering long-term results. The modified Ravitch technique is more
adequate as it can be used in all types of deformities and in
concomitant surgery allowing optimal operative exposure during cardiac
procedures, easy postoperative reentry and resuscitation maneuvers if
needed.
References:
1- Hysi I, Vincentelli A, Juthier F, et al: Cardiac surgery and repair
of pectus deformities: When and how? Int J Cardiol. 194:83-6, 2015
2- Kara M, Gundogdu AG, Kadioglu SZ, Cayirci EC, Taskin N: The use of
sternal wedge osteotomy in pectus surgery: when is it necessary? Asian
Cardiovasc Thorac Ann. 24(7):658-62, 2016
3- Kim SY, Park S, Kim ER, et al: A Case of Successful Surgical Repair for Pectus Arcuatum Using
Chondrosternoplasty. Korean J Thorac Cardiovasc Surg. 49(3):214-7, 2016
4- Leng S, Bici K, Facchini F, et al: Customized Cutting Template to
Assist Sternotomy in Pectus Arcuatum. Ann Thorac Surg.
107(4):1253-1258, 2019
5- Kuzmichev V, Ershova K, Adamyan R: Surgical correction of pectus arcuatum. J Vis Surg. 2:55, 2016
6- Emil S: Current Options for the Treatment of Pectus Carinatum: When
to Brace and When to Operate? Eur J Pediatr Surg. 28(4):347-354, 2018
Imposter Syndrome
Imposter syndrome refers to a feeling of self-doubt or
innate fear of being discovered as a fraud or non-deserving
professional, despite their demonstrated talent and achievements.
Imposter syndrome is characterized by a chronic sense of self-doubt
coupled with a constant worry of being discovered as a fraud. Imposter
syndrome is more prevalent in high achievers, women, and
under-represented racial, ethnic, and religious minorities. Impostor
syndrome is increasingly recognized as a condition between physicians
and physicians in training. Despite remarkable academic and
professional achievements affected individuals beliefs that they
were unintelligent. For affected individuals, imposter syndrome can
lead to burnout, psychological distress, emotional suffering, and
serious mental health disorders, including chronic dysphoric stress,
anxiety, depression, drug abuse and suicide. Most cases start early
during high school or college. The true incidence of imposter syndrome
is unknown in medical professionals. In the US, among medical students
the rate of imposter syndrome was 49% in women and 24% in men, and
among residents the rate was similar. The root cause of imposter
syndrome is not known, but it has been related to depression and
anxiety, which are both present among residents, with suicide being the
second most common cause of death among residents. Imposter syndrome
among general surgery residents is not only prevalent but severe with
76% of residents reporting either significant or severe imposter
syndrome. Neither sex nor age correlates with the presence of, or level
of, imposter syndrome in the general surgery resident population. It is
believed that imposter syndrome is an incidentally protective mechanism
encouraged by the hierarchical culture of surgical training by which
residents are encouraged to self-regulate their decision-making
process. By constantly downplaying their own accomplishments, those
suffering from imposter syndrome may sabotage their own career.
Institutions must address imposter syndrome by increasing the
visibility of the problem, providing access to mental health coaching,
and establishing supportive organization policies. Institutions and
residency programs should provide training for mentors to help them
recognize the negative consequence of the imposter syndrome. Medical
educators must recognize that it is not just the underperforming or
failing learners who struggle and require support, and medical culture
must create space for physicians to share their struggles. The
Accreditation Council for Graduate Medical Education (ACGME) requires
residency programs to support residents well-being via
established policies and programs. Imposter syndrome has been linked to
burnout and suicide in residents and understanding how to combat it may
help improve resiliency in residents. Imposter syndrome has been linked
to resident burnout and discussing imposter syndrome is viewed as an
effective intervention to promote resident wellness and resiliency.
When creating wellness interventions, residency programs should
consider addressing imposter syndrome.
References:
1- Kimyon RS: Imposter Syndrome. AMA J Ethics 22(7): E628-629, 2020
2- Mullangi S, Jagsi R: Imposter Syndrome: Treat the Cause, Not the Symptom. JAMA. 322(5):403-404, 2019
3- Chrousos GP, Mentis AA: Imposter syndrome threatens diversity. Science. 367(6479):749-750, 2020
4- Baumann N, Faulk C, Vanderlan J, Chen J, Bhayani RK: Small-Group
Discussion Sessions on Imposter Syndrome. MedEdPORTAL. 16:11004, 2020
5- Bhama AR, Ritz EM, Anand RJ, et al: Imposter Syndrome in Surgical
Trainees: Clance Imposter Phenomenon Scale Assessment in General
Surgery Residents. J Am Coll Surg. 233(5):633-638, 2021
6- Gottlieb M, Chung A, Battaglioli N, Sebok-Syer SS, Kalantari A:
Impostor syndrome among physicians and physicians in training: A
scoping review. Med Educ. 54(2):116-124, 2020
Short Bowel Syndrome
Short bowel syndrome (SBS) refers to a compromised bowel
absorptive capacity due to severely reduced mucosal surface resulting
in diarrhea, water-electrolytes imbalances, and protein malnutrition.
SBS is the most common cause of intestinal failure in children. Most
underlying conditions that lead to major loss of intestine in neonates
have their origin in intrauterine life. SBS usually occurs after
extensive bowel resection, either congenital or acquired, such as that
associated with small bowel atresia, complex gastroschisis, midgut
volvulus and necrotizing enterocolitis. The most common acquired cause
of SBS is NEC with 30% in most reported series. Factors influencing
outcome in SBS include underlying diagnosis, type of segments
preserved, stoma vs primary anastomosis, presence of ileocecal valve
and the age of the child at the time of surgery. Massive
resection stimulates modification in thickness and length of the muscle
layer and villi crypts. Distension of the remaining bowel is the most
common consequence after massive resection. Massive jejunal resections
are better tolerated than significant ileal resections. Ileal
resections are associated with impaired resorption of Vitamin 12, bile
salts and fatty acids. Three anatomical subtypes of SBS: (1) small
bowel resection with anastomosis and intact colon; (2) small bowel
resection with partial colon resection; (3) small bowel resection with
high output jejunostomy. Type 1 has the best potential for adaptation,
while type 3 the least. With the advent of parenteral nutrition support
survival of SBS improved significantly. Management of SBS aims to
promote adaptation of the remnant bowel. Parenteral nutrition (PN)
provides nutrition while the bowel achieves intestinal autonomy. The
bowel should be use for feeding as much and early as possible to
stimulate adaptation. Oral feeding maintains sucking and swallowing
functions, promotes release of epidermal growth factor from salivary
glands and increases GI secretion of trophic factors. Breast feeding
should be encouraged. Long term PN leads to sepsis, cholestasis due to
liver failure and death. Key predictors of mortality in SBS include
cholestasis (conjugated bilirubin > 2.5 mg%) and percentage of small
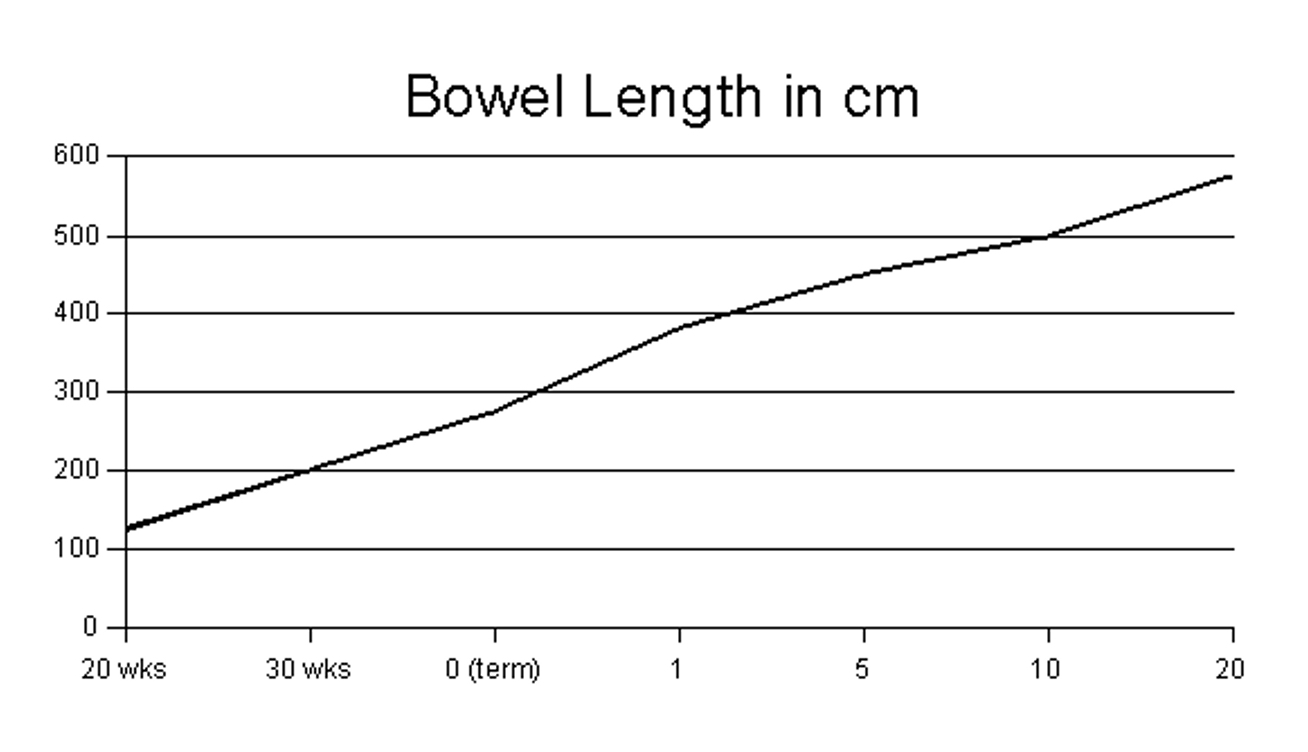
bowel length. A small bowel length greater than 10% of expected for a
given gestational age is highly predictive of survival (see normal
bowel length in accordance with gestational age graph). Presence of an
ileocecal valve and percentage of small bowel length are primary
predictors of weaning PN. Surgical approaches to maximize bowel
digestive and absorptive function are important in the management of
SBS. These include stoma closure, bowel continuity restoration,
resection of strictures and closure of fistula. When the bowel is
short, dilated, and static children might benefit from longitudinal
intestinal lengthening and tapering (Bianchi) or serial transverse
enteroplasty (STEP) procedures. The UGIS provide accurate estimates of
bowel diameter and length use to operative planning. Surgical bowel
lengthening should be considered in any chronically PN-dependent child
when there is substantial bowel dilatation and symptoms of small
intestinal bacterial overgrowth regardless of remaining bowel length.
Medical approach to SBS include antidiarrhea/ antimotility agents and
controlling acid/base balance. Hypersecretion of gastrin and gastric
acid occurs in children after extensive small bowel resection which
should be managed. Octreotide inhibits gastrin and diarrhea prolonging
transit time. Intestinal bacterial overgrowth frequently seen in SBS is
managed with probiotic and antibiotic therapy. Promising hormonal
therapy include glucagon-like peptide 2 hormone (Teduglutide) produced
by the L-cells of the terminal ileum. Teduglutide has a trophic effect
on the bowel, promotes absorption and adaptation. The future of SBS
might lie in an artificial grown and engineered harvested intestine.
Micronutrient deficiencies are frequent during intestinal
rehabilitation for SBS. The most common micronutrient deficiency
include zinc, copper, vitamin D and phosphorus after the transition to
enteral nutrition. With liver failure and reduced venous access, bowel
transplantation becomes the treatment of choice.
References:
1- Goulet O, Finkel Y, Kolacek S, Puntis J: Chapter 5.2.1. Short Bowel
Syndrome: Half a Century of Progress. J Pediatr Gastroenterol Nutr. 66
Suppl 1:S71-S76, 2018
2- Spencer AU, Neaga A, West B, et al: Pediatric short bowel syndrome:
redefining predictors of success. Ann Surg. 242(3):403-9, 2005
3- Coletta R, Khalil BA, Morabito A: Short bowel syndrome in children:
surgical and medical perspectives. Semin Pediatr Surg. 23(5):291-7, 2014
4- Mutanen A, Wales PW: Etiology and prognosis of pediatric short bowel syndrome. Semin Pediatr Surg. 27(4):209-217, 2018
5- Hill S, Carter BA, Cohran V, et al: Safety Findings in Pediatric
Patients During Long-Term Treatment With Teduglutide for Short-Bowel
Syndrome-Associated Intestinal Failure: Pooled Analysis of 4 Clinical
Studies. JPEN J Parenter Enteral Nutr. 45(7):1456-1465, 2021
6- Hollwarth ME: Surgical strategies in short bowel syndrome. Pediatr Surg Int. 2017 33(4):413-419, 2017
7- Feng H, Zhang T, Yan W, et al: Micronutrient deficiencies in
pediatric short bowel syndrome: a 10-year review from an intestinal
rehabilitation center in China. Pediatr Surg Int. 36(12):1481-1487, 2020
PSU Volume 58 No 03 MARCH 2022
Extremity Compartment Syndrome
Extremity compartment syndrome (ECS) in children is a
potential cause of permanent disability. Sustained increased pressure
in the limb fascial compartments compromises circulation causing
ischemia and necrosis of the contents within. Early recognition is
critical in avoiding further disability. Diagnosis of compartment
syndrome in children can be challenging due to poor cooperation,
difficulty with communication and difficulty measuring compartment
pressures of the affected limb in the conscious child. The most common
risk factors for ECS in the pediatric age comprise tibial diaphysis
fractures, soft-tissue injury, distal radius fracture, radius and ulna
diaphysis fracture and crush injury. Extremity fractures causes most
ECS in children (75%) followed by vascular injury, tibial osteotomy,
and soft-tissue injury. ECS can also arise from extrinsic causes that
exert pressure such as compressive casts or bandages, pneumatic
antishock garments, or intrinsic factors that increase the volume
inside the fascial envelops such as septic arthritis, intraosseous
infusions, toxic venom, burns, intramuscular hematomas, hereditary
bleeding disorders and viral diseases. An open fracture of the forearm
or leg significantly increases the risk for ECS. Nontraumatic causes of
limb compartment syndrome in children include ischemia-reperfusion
events after arterial injury, thrombosis, burns, bleeding disorders and
blunt injury. The pathogenesis of ECS is tissue damage leading to
increased intracompartmental pressure way above the closing pressure of
venules. Continued arterial inflow increases the pressure until the
arterioles develop stasis and ischemia occurs. Prolonged ischemia
beyond a six-hours period results in ischemic muscle which may result
in myonecrosis, chronic contracture and permanent nerve damage.
Compartment syndrome is a clinical diagnosis. The affected patient
develops paresthesia, numbness, swelling and pain out of proportion or
with passive movement of the extremity. Diminished pulses, pallor and
progressive neurologic deficit are late findings less commonly seen.
Pain is one of the earliest symptoms of ECS. Sensory deficit occurs
before motor dysfunction. Paresthesia in the affected extremity is one
of the first signs of hypoxia to nerve tissue within a compartment.
Blood flow in the capillary circulation ceases when compartment
pressure exceeds 35 mm Hg. The sensory nerves are affected first,
followed by the motor nerves and muscle, fat and skin become involved
later. ECS can be confirmed by the measurement of tissue compartment
pressure greater than 30 mm of Hg. The normal pressure in a muscle
compartment is less than 10-12 mm Hg. This diagnostic method is
essential in uncooperative, altered mental status, very young or
children with inconsistent clinical symptoms. Measurement of
compartment pressure can be performed using a slit catheter, wick
catheter, needle manometer, electronic arterial pressure transducer, or
a solid-state transducer intra compartment catheter. Management of
symptomatic ECS with pressures above 30 mm Hg is urgent decompressive
fasciotomy. Favorable outcomes are found in children who had a
fasciotomy less than six hours from the time of diagnosis. After
fasciotomy, limb compartment pressures should be monitored as
progressive muscle swelling may continue as a result of toxic effects
of infection. The lower leg is the most common location of acute ECS
with the anterior and lateral compartments most frequently affected.
Children tolerate increased intracompartmental pressure for longer
periods of time than adults before tissue necrosis becomes
irreversible. The most common complications after ECS in children is an
unpleasant scar since wound closure after upper or lower extremity
fasciotomies require split thickness skin graft. Silent compartment
syndrome is defined as confirmed compartment syndrome without
significant pain or absence of marked pain on passive motion. Pediatric
patients generally achieve good outcomes even when presenting in a
delayed fashion and undergoing fasciotomies after 24 hours of the
initial event. Decompressive fasciotomy is recommended even if there is
prolonged time from injury to diagnosis.
References:
1- Grottkau BE, Epps HR, Di Scala C: Compartment syndrome in children and adolescents. J Pediatr Surg. 40(4):678-82, 2005
2- Kanj WW, Gunderson MA, Carrigan RB, Sankar WN: Acute compartment
syndrome of the upper extremity in children: diagnosis, management, and
outcomes. J Child Orthop. 7(3):225-33, 2013
3- von Keudell AG, Weaver MJ, Appleton PT, et al: Diagnosis and
treatment of acute extremity compartment syndrome. Lancet.
386(10000):1299-1310, 2015
4- Shirley ED, Mai V, Neal KM, Kiebzak GM: Wound closure expectations
after fasciotomy for paediatric compartment syndrome. J Child
Orthop. 12(1):9-14, 2018
5- Frei B, Sommer-Joergensen V, Holland-Cunz S, Mayr J: Acute
compartment syndrome in children; beware of "silent" compartment
syndrome: A CARE-compliant case report. Medicine (Baltimore).
99(23):e20504, 2020
6- Lin JS, Samora JB: Pediatric acute compartment syndrome: a
systematic review and meta-analysis. J Pediatr Orthop B. 29(1):90-96,
2020
Diverticulitis in Children
The most common diverticulum in children causing surgical
problems is the Meckel's diverticulum. In very rare occasion the
pediatric patient can develop diverticular disease of the colon similar
to that occurring in the adult. Such diverticular disease can lead to
colonic diverticulitis. Diverticulitis is predominantly a disease of
adults older than 50 years of age, being extremely rare in children.
Low quantity dietary fiber, obesity, constipation, decreased physical
activity, steroids, and smoking all predispose individuals to
diverticulosis. Chronic increase intraluminal pressure leads to
formation of pseudo-diverticular outpouching. Less than 5% of all
patients with diverticula of any etiology develop diverticulitis. The
prevalence in the population younger than 40 is only 10%. No matter the
age group or etiology, diverticula can develop anywhere along the colon
from the cecum and appendix to the sigmoid colon. When diverticular
disease occurs in children, they are associated with alterations in the
component of the colonic wall. Some genetic disorders in children are
associated with diverticulitis due to weakening of the colonic wall by
alteration of collagen or elastin synthesis within the tissues. They
include cystic fibrosis, Ehlers-Danlos syndrome, Marfan syndrome and
William-Beuren syndrome. Possible complications of colonic
diverticulosis include bleeding, inflammation (diverticulitis), and
perforation. Symptoms depend on localization of the diverticula.
Differential diagnosis includes colon cancer, Crohn's disease, ischemic
colitis, pseudomembranous enterocolitis, and pelvic inflammatory
disease. CT-Scan or MRI is utilized to diagnose diverticulitis.
Ultrasound and MRI can be useful alternatives in the initial evaluation
of a patient with suspected acute diverticulitis when CT imaging is not
available or is contraindicated. Pediatric colonic diverticulitis is
often associated with a more complicated course than that seen in
adults patients. Most pediatric cases have been described in the
cecum or ascending colon as a true diverticulum. Right sided
diverticulitis is relatively rare inflammatory condition affecting the
cecum and ascending colon. The incidence in children has not been
determined since most cases are incorporated into adult series. They
may be found as solitary lesion, multiple lesions, or parts of
generalized diverticulosis of the colon. Right colonic diverticula are
predominantly congenital and solitary being true diverticula consisting
of all layers of the bowel wall. Most children complain of right lower
quadrant pain with tenderness associated with nausea and vomiting.
Since they can mimic appendicitis, the diagnosis is often difficult.
Nonoperative management with antibiotics and bowel rest is advocated by
most, leaving resection or diverticulectomy for recurrent episodes,
obstructing mass, abscess, fistula, or perforation. The recurrence rate
of children managed successfully with intravenous antibiotics is 18%.
Classic findings related to sigmoid diverticulitis in adults include
left lower quadrant pain, fever, and leukocytosis. Complicated
diverticulitis is defined as diverticulitis associated with
uncontained, free perforation with systemic inflammatory response,
fistula, abscess, stricture, or obstruction. Micro-perforation in the
absence of a systemic inflammatory response is not considered
complicated diverticulitis. Symptomatic uncomplicated disease is
defined as diverticulosis with associated chronic abdominal pain in the
absence of clinically overt colitis. CRP above 150 mg/L is associated
with complicated diverticulitis. Elevated procalcitonin is associated
with diverticulitis recurrence. Most cases resolved with
antibiotics. Patients with recurrent symptoms, diverticular abscess,
fistula, obstruction, or stricture will need surgery. An elective
resection based on young age at presentation is not recommended.
Management of pediatric patients with diverticulitis should be
multidisciplinary, including GI, surgery, genetics, cardiology, and
ophthalmology.
References:
1- Santin BJ, Prasad V, Caniano DA: Colonic diverticulitis in
adolescents: an index case and associated syndromes. Pediatr Surg Int.
25(10):901-5, 2009
2- Ignacio RC Jr, Klapheke WP, Stephen T, Bond S: Diverticulitis in a
child with Williams syndrome: a case report and review of the
literature. J Pediatr Surg. 47(9):E33-5, 2012
3- Yano K, Muraji T, Hijikuro K, Shigeta K, Ieiri S: Cecal diverticulitis: Two pediatric cases. Pediatr Int. 61(9):931-933, 2019
4- Hall J, Hardiman K, Lee S, et al: The American Society of Colon and
Rectal Surgeons Clinical Practice Guidelines for the Treatment of
Left-Sided Colonic Diverticulitis. Dis Coln Rectum 63: 728-747, 2020
5- Cadiz EM, Doan S, Kruszewski P: An Unusual Case of Diverticulitis in
a Teenager With a Complicated Clinical Course. J Pediatr Gastroenterol
Nutr. 71(5):e142, 2020
6- Lee ZW, Albright EA, Brown BP, Markel TA: Congenital cecal
diverticulitis in a pediatric patient. J Pediatr Surg Case Rep. 2021
Sep;72:101929. doi: 10.1016/j.epsc.2021.101929.
Epub 2021 Jun 2.
7- Hatakeyama T, Okata Y, Miyauchi H, et al: Colonic diverticulitis in
children: A retrospective study of 16 patients. Pediatr Int.
63(12):1510-1513, 2021
Inguinal Lymphadenopathy
Inguinal lymphadenopathy (IL) refers to the condition in
which peripheral inguinal or groin lymph nodes become abnormally
enlarged, sometimes tender to palpation causing concern to caretakers.
Lymphadenopathy is a common clinical manifestation in the pediatric age
group. It may be part of normal age-related physiology or in response
to any local or generalized infection in the body. Inguinal
adenopathy may be a symptom to several disease process, most commonly
of infectious origin. The history of the child with IL should be
studied carefully since it provides clues to the underlying disease.
Most groin adenopathies are self-limited infections in young patients.
When confronted with a child with IL laboratory tests, imaging studies
and tissue diagnosis might be needed unless there is a clear
explanation for the sudden growth of the lymph node. Ultrasound, a
non-invasive and non-radiating imaging study, is the best study to
assess lymph nodes in the groin and neck. CT-Scan should be avoided
with peripheral lymphadenopathies to reduce radiation injury associated
with this imaging modality. CT-Scan is more helpful with central lymph
nodes in the thorax or abdominopelvic cavities. Fine needle aspiration
(FNA) biopsy can be used as initial management. FNA biopsy is
easy, safe, rapid and a cost-effective tool, but will need a
cooperating child to be performed. Excisional biopsy of the enlarged
lymph node is the gold standard procedure and in the groin the
procedure is usually simple, fast, and free from major complications.
In fact, the most common complication is seroma which resolves
spontaneously in most cases. In the groin lymph nodes larger than 1.5
cm in children are abnormal. It must be determined if the
lymphadenopathy is localized or generalized. Generalized
lymphadenopathy is defined as the enlargement of two or more groups of
noncontinuous groups of lymph nodes. It results from systemic illness
like infections (viral, bacterial, fungal, and protozoan),
malignancies, autoimmune disease, drugs reactions, histiocytic
disorders, disseminated neoplastic diseases and storage disorders. If
the child does not present with overt signs of malignancy, the lymph
node can be safely watch for three to four weeks before considering
biopsy. Benign lesions are more commonly encountered than malignant
lesions in children and include benign reactive hyperplasia (by far the
most common pathology; approximately 65-85%), chronic skin diseases,
cat-scratch disease, toxoplasmosis, necrotizing granulomatosis, and
non-necrotizing granulomatosis. Of the malignant conditions,
non-Hodgkin lymphoma is the most common. FNA biopsy can be helpful in
differentiating benign from malignant pathology but is often faced with
failure due to quantity of tissue to provide a diagnosis. As mentioned
previously, an open biopsy with removal of the whole adenopathy is
almost always diagnostic. Chronic skin disorders are a cause of what is
known as dermatopathic lymphadenopathy, an entity that represents a
secondary immune response to a pathologic condition affecting primarily
the skin.
References:
1- Ozkan EA, Goret CC, Ozdemir ZT, et al: Evaluation of peripheral
lymphadenopathy with excisional biopsy: six-year experience. Int J Clin
Exp Pathol. 8(11):15234-9, 2015
2- Garces S, Yin CC, Miranda RN, et al: Clinical, histopathologic, and
immunoarchitectural features of dermatopathic lymphadenopathy: an
update. Mod Pathol. 33(6):1104-1121, 2020
3- Khatri A, Mahajan N, Malik S, Rastogi K, Kumar P, Saikia D:
Peripheral Lymphadenopathy in Children: Cytomorphological Spectrum and
Interesting Diagnoses. Turk Patoloji Derg. 37(3):219-225, 2021
4- Sandoval AC, Reyes FT, Prado MA, Pena AL, Viviani TN: Cat-scratch
Disease in the Pediatric Population: 6 Years of Evaluation and
Follow-up in a Public Hospital in Chile. Pediatr Infect Dis J.
39(10):889-893, 2020
5- Abantanga FA: Groin and scrotal swellings in children aged 5 years and below: a review of 535
cases. Pediatr Surg Int. 19(6):446-50, 2003
6- Hasanbegovic E, Mehadzic S: [Etiology of lymphadenopathy in childhood]. Med Glas (Zenica). 7(2):132-6, 2010
PSU Volume 58 NO 04 APRIL 2022
Brunner Gland Adenoma
Brunner gland adenoma (BGA), also known as an hamartoma, is
a rare duodenal lesion comprising not more than 5% of benign duodenal
lesions. Most Brunner gland adenomas are of small size without causing
significant symptoms. When BGA grows, they can cause obstruction or
bleeding. Brunner glands are submucosal mucin-secreting glands
predominantly located in the posterior wall of the duodenal bulb and
second portion of the duodenum segment and progressively decrease in
size and number in the distal potions of the proximal bowel. Brunner's
glands secrete an alkaline fluid composed of viscous mucin which
protects the duodenal epithelium from acid chyme of the stomach. BGA
are rarely larger than 2 cm in asymptomatic individuals, while larger
than 5 cm in symptomatic patients. Brunner's gland could be classified
into three types based on size: type 1 (diffuse nodular hyperplasia)
confined to the mucosa with multiple sessile projections occupying most
of the duodenum; type 2 (circumscript nodular hyperplasia), found in
the bulb duodenum and usually smaller than 1 cm; and type 3 (Brunner's
gland adenoma) stemmed with sized 1-2 cm, generally without clinical
manifestations. Etiology of BGA is unknown. It is believed that gastric
hypersecretion of acid results in hyperplasia of Brunner's gland
resulting in adenoma formation and excrescence toward the bowel lumen.
Others believe the loss of the alkaline protection of the exocrine
pancreas leads to compensatory hyperplasia of Brunner's gland with
increased production of mucus and alkali. Helicobacter infection has
also been found culprit of BGA, though a clear relationship has not
been met. It is believed BGA are hamartomas. The word adenoma might be
a misnomer, since the mass is not a true neoplasm, but rather a
hamartomatous or hyperplastic collection of mature glands with no known
potential for malignant transformation. Patients with BGA are usually
asymptomatic, or developed symptoms of nausea, vomiting, bloating,
dyspepsia, vague abdominal pain, melena, or hematemesis. Pancreatitis
(ampullary lesions), intussusception and diarrhea have also been
reported. Those in the pylorus often presents with epigastric pain,
dyspnea, or melena, whereas those in the posterior wall of duodenum
often presents with postprandial fullness. When the adenoma grows it
can cause symptoms of obstruction or gastrointestinal bleeding. Chronic
bleeding with ulceration is found in most symptomatic patients.
On rare occasion BGA can cause gastric outlet obstruction. The most
common laboratory finding in symptomatic patients is anemia. Abdominal
imaging (Ultrasound/CT-Scan/MRI) or endoscopy can detect the lesion.
Upper endoscopy localizes the lesion and biopsy can provide the
diagnosis of a BGA. Endoscopic biopsy shows involvement of the mucosa
and submucosa layers without deeper extension, variable echogenicity,
and cystic changes within the lesion. Cytologically, Brunner's gland
shows loose clusters of flat, two-dimensional cells with minimal
overlapping or atypia showing abundant, finely granular, and vacuolated
cytoplasm. In most cases, diagnosis is confirmed after endoscopic or
surgical resection. Endoscopic resection is recommended to avoid
developing symptoms with time. When endoscopic resection is not
possible, surgical resection is indicated. Recurrence rate after both
modalities of management is very low. BGA have been reported very
rarely in the pediatric age and can be associated with surgical repair
of duodenal atresia. BGA are benign and the prognosis after endoscopic
or surgical management is good.
References:
1- Lu L, Li R, Zhang G, Zhao Z, Fu W, Li W: Brunner's gland adenoma of
duodenum: report of two cases. Int J Clin Exp Pathol. 8(6):7565-9, 2015
2- Marinacci LX, Manian FA: Brunner Gland Adenoma. Mayo Clin Proc. 92(11):1737-1738, 2017
3- Ortiz Requena D, Rojas C, Garcia-Buitrago M: Cytological diagnosis
of Brunner's gland adenoma (hyperplasia): A diagnostic challenge. Diagn
Cytopathol. 49(6):E222-E225, 2020
4- Liang M, Liwen Z, Jianguo S, Juan D, Ting S, Jianping C: A case
report of endoscopic resection for the treatment of duodenal Brunner's
gland adenoma with upper gastrointestinal bleeding. Medicine
(Baltimore). 99(52):e23047, 2020
5- Yang B, Li K, Luo R, Xiong Z, Wang L, Xu J, Fang D: Large Brunner's
gland adenoma of the duodenum for almost 10 years. Open Life Sci.
15(1):237-240, 2020
6- Zhou SR, Ullah S, Liu YY, Liu BR: Brunner's gland adenoma: Lessons learned for 48 cases. Dig Liver Dis. 53(1):134-136, 2021
Hirschsprung's Associated Enterocolitis (HAEC)
Hirschsprung's disease (HD) occurs in approximately one in
5000 live births with most babies presenting with failure to pass
meconium in the first 24 hours of life. Failure to recognize HD early
in infancy place them at high risk of developing HirschsprungÕs
associated enterocolitis (HAEC). HAEC is a serious life-threatening
inflammatory complication of HD. HAEC occurs preoperatively in 6-26% of
cases, and post pull-through surgery in 5-42%. HAEC is histologically
characterized by cryptitis, the appearance of neutrophils in intestinal
crypts. They progress to crypt abscess, mucosal ulceration and
fibrinopurulent debris. In severe cases ischemia, transmural necrosis
and perforation can occur leading to shock and hypoperfusion.
Pathophysiologic mechanism associated with HAEC includes partial
mechanical obstruction (bacterial translocation), unbalanced microflora
(dysbiosis), insufficient immunoglobulin secretion, abnormal mucin
production (impaired mucosal barrier), and dysfunction of the enteric
nervous system. Mortality of HAEC ranges between 1% and 10%. Several
factors contribute to an increased risk of HAEC, these include family
history of HD, Down syndrome, long-segment HD involvement, obstruction
from any cause (retained aganglionosis, transitional zone pull-through,
dysmotility following pull-through, anastomotic stricture, twist in the
pull-through or tight muscular cuff following surgery), and prior
episodes of HAEC. Some authors believe no patient or clinical
characteristic is associated with the risk of postoperative HAEC. The
risk of developing HAEC increases with the length of the aganglionic
segment and delay in diagnosis. The diagnosis is suspected when a child
with constipation develops abdominal distension, pain, vomiting,
explosive watery diarrhea, fever, lethargy, rectal bleeding, and shock.
Initial images consist of plain radiographs which can demonstrate a
cutoff sign in the rectosigmoid colon with absent distal air, dilated
proximal bowel loops, air fluid levels, pneumatosis intestinalis,
"sawtooth' appearance with irregular intestinal lining, or
pneumoperitoneum from bowel perforation. Barium enema and colonoscopy
are contraindicated in the acute setting due to the risk of
perforation, while CT-Scans are of little value in the diagnosis and
treatment of HAEC. Chronic HAEC symptoms include persistent diarrhea,
soiling, intermittent abdominal distension, and failure to thrive.
Should this occur after surgical management mechanical obstruction from
aganglionosis should be suspected and confirmed with rectal biopsy. The
diagnosis of HD is made histologically by the absence of ganglion
cells, presence of nerve hypertrophy and absent calretinin
immunohistochemistry. For prophylactic prevention of HAEC routine
rectal irrigation or diverting colostomy is indicated in selected
patients. Rectal irrigations reduce fecal stasis and bacterial load,
limiting colon distension. Children with HD and severe congenital heart
disease should be diverted to avoid HAEC. Probiotics management has
controversial results as a preventive measure. The clinical suspicion
and severity of HAEC have been graded based on history, physical
examination, imaging studies and laboratory findings similar to Bell's
criteria for neonatal enterocolitis. Four or more of 16 criteria seen
in the Table below are diagnostic. Such score helps make the correct
diagnosis of HAEC. Existing scoring systems perform poorly in
identifying episodes of HAEC, resulting in significant underdiagnosis.
Management of HAEC includes fluid resuscitation using isotonic
solutions, broad spectrum intravenous antibiotics, decompression of the
gastrointestinal tract and bowel rest. Systemic antibiotics are used
empirically in HAEC, with metronidazole typically chosen to managed
anaerobes. Those children with severe sepsis and acutely ill (Grade
III) benefit with admission to the intensive care unit some of them
needing vasopressor therapy and ventilatory support. Rectal washouts
with warm saline are the mainstem of management and they should be
performed two to four times daily at a rate of 10-20 ml/kg of weight
each time or until the effluent is clear. In newborns presenting with
severe HAEC, shock and sepsis immediate diversion (leveling colostomy)
should be considered. Other risk factor associated with the need of
diversion includes delayed presentation, co-morbid conditions, presence
of HAEC or multiple risk factors for HAEC in the preoperative period.
Diversion improves patient symptoms but does not resolve HAEC
development later in life. HAEC can also occur in the postoperative
period after pull-through definitive surgery. Rectal washouts are also
effective to manage postoperative HAEC. There is a trend toward a
higher incidence of enterocolitis in the primary endorectal
pull-through group as compared with those with a two-stage approach.
Postoperative HAEC can occur more than 18 months after definitive
surgery. The postoperative risk of developing HAEC after
definitive surgery is highest in those operated with Swenson and
Duhamel approach (20%), and lowest with Soave procedure (11%). Children
with recurrent HAEC, occurring in 2-33% of patients, should be
evaluated for anatomic or pathologic causes of obstruction. A
water-soluble contrast enema can identify any mechanical cause of
obstruction. Should the suspicion of persistent aganglionosis be
considered a rectal biopsy should be performed. Risk factors for
recurrent HAEC includes preoperative HAEC, history of central nervous
system infection and congenital chromosome anomalies and does not
include placement of an ostomy prior to pull-through and congenital
cardiac anomalies. Recurrent postoperative HAEC does not have an impact
on mortality. Children with previous episodes of HAEC are more likely
to develop subsequent episodes.
References:
1- Frykman PK, Short SS: Hirschsprung-associated enterocolitis: prevention and therapy. Semin Pediatr Surg. 21(4):328-35, 2012
2- Demehri FR, Halaweish IF, Coran AG, Teitelbaum DH:
Hirschsprung-associated enterocolitis: pathogenesis, treatment and
prevention. Pediatr Surg Int. 29(9):873-81, 2013
3- Gosain A, Frykman PK, Cowles RA, et al: Guidelines for the diagnosis
and management of Hirschsprung-associated enterocolitis. Pediatr Surg
Int. 33(5):517-521, 2017
4- Frykman PK, Kim S, Wester T, et al: Critical evaluation of the
Hirschsprung-associated enterocolitis (HAEC) score: A multicenter study
of 116 children with Hirschsprung disease. J Pediatr Surg.
53(4):708-717, 2018
5- Pruitt LCC, Skarda DE, Rollins MD, Bucher BT:
Hirschsprung-associated enterocolitis in children treated at US
children's hospitals. J Pediatr Surg. 55(3):535-540, 2020
6- Roorda D, Oosterlaan J, van Heurn E, Derikx JPM: Risk factors for
enterocolitis in patients with Hirschsprung disease: A retrospective
observational study. J Pediatr Surg. 56(10):1791-1798, 2021
7- Lewit RA, Veras LV, Cowles RA, et al: Reducing Underdiagnosis of
Hirschsprung-Associated Enterocolitis: A Novel Scoring System. J Surg
Res. 261:253-260, 2021
8- Hagens J, Reinshagen K, Tomuschat C: Prevalence of
Hirschsprung-associated enterocolitis in patients with Hirschsprung
disease. Pediatr Surg Int. 38(1):3-24, 2022
Transanal Pull-Through
In 1998, De la Torre introduced a new one-stage surgical
technique to manage Hirschsprung's disease (HD) where rectal
mucosectomy, aganglionic segment colectomy, posterior myectomy, and
normoganglionic colon pull-through is performed through the anus. HD
aganglionosis affects the rectosigmoid (classic HD; 85%), long-segment
(10%) and total aganglionosis (5%) of the colon with sporadic cases
affecting the proximal small bowel. The transanal pull-through (TAPT)
is mainly design for children with classic HD and uses a prone
approach, though it can also be performed in supine position.
Advantages of the TAPT include abdominal or intraperitoneal bowel
opening is not necessary unless the child has a previous leveling
colostomy, risk of adhesion is decreased, excellent cosmetic results,
earlier full oral feedings, shorter hospital stay, less costs, less
pain, reduced operating time, endorectal dissection preserves the
anorectal sphincters and their blood supply along with innervation
causing less damage to fecal and urinary incontinence. Should the
ganglionic bowel need further dissection proximally laparoscopy can
provide this mobilization. The procedure can be performed in newborns
with potential benefits of avoiding a colostomy and establishing
colonic continuity early in life improving continence results. A stoma
in HD is considered appropriate in children with bowel perforation
(usually cecal), severe malnutrition, severe enterocolitis, very
dilated proximal bowel, total colonic aganglionosis or lack of adequate
pathological support. Should the surgeon choose to wait beyond the
neonatal period for final correction of HD the risk of developing
enterocolitis should be minimized by ensuring adequate decompression of
the proximal dilated bowel by appropriate daily irrigations,
administration of prophylactic metronidazole or probiotics, and use of
breast feeding or elemental infant formula. Using laparoscopy
concomitantly with the transanal approach can help in the proximal
bowel dissection, distal bowel dissection and obtaining a seromuscular
biopsy for frozen section to determine the ganglionic bowel to be
pull-through. A frozen full-thickness biopsy helps the pathologist see
both submucosa and myenteric plexus in look for ganglion cell and nerve
hypertrophy minimizing error. Whether using a supine or prone position
for the procedure depends on the certainty that you are dealing with
classic HD and proximal mobilization will not be required. The prone
position has the advantage that mesenteric vessels can be send and
controlled more effectively than in the supine or lithotomy position.
The transanal dissection can be started 0.5-1 cm above dentate line in
neonates and 1-2 cm above the dentate line in older children as the
transitional epithelium must not be damaged to avoid loss of sensation
and incontinence. Dissection on the outside of the rectum, as that used
during laparoscopic or open Swenson procedures, may increase the risk
of injury to pelvic nerves and vessels, and to the prostate, urethra,
and vagina. Comparison between complete TAPT and laparoscopic-assisted
TAPT do not differ between rates of major complications, including
leaks, strictures, enterocolitis, fecal incontinence, postoperative
obstructive symptoms, and mean length of stay. Comparison between the
Duhamel procedure and the TAPT have shown they are similar in respect
postoperative fecal incontinence and operation time, but the Duhamel is
associated with longer hospital stay and lower rate of enterocolitis.
When performing the TAPT in neonates, a higher incidence of
enterocolitis has been reported due to increased risk of sphincter
spasm and anastomotic strictures. Excising the entire posterior rectal
muscle cuff (myectomy) has been effective in reducing the incidence of
enterocolitis. Current guidelines suggest doing the TAPT between two
and three months of age if the child is growing well and the bowel is
sufficiently decompressed. Whether performing the procedure totally
transanally or laparoscopy assisted does not affect long-term bowel
function. More than 20% of patients develop at least one complication
within the 30-days following a TAPT. Older age at time of surgery
increases the risk of developing a postoperative complication. They
include anastomotic leakage, abdominal abscess, and anastomotic
strictures. Ischemia and increased tension on the colon anastomosis
play an important role in leakage and stricture. This is one reason to
consider perioperative diverting stoma in older patients. Older age at
time of surgery, laparotomy-assisted and long segment disease increases
the risk of developing a postoperative complication. Long term problems
with pull-through surgery for HD include obstructive symptoms (30%),
persistent constipation, soiling, enterocolitis and descending
aganglionic bowel. Persistent bowel obstructive problems might be
caused by mechanical obstruction, recurrent or acquired aganglionosis,
disordered motility in the proximal colon, nonrelaxation of the
internal anal sphincter, or stool-holding behavior. Mechanical
obstruction might also be caused by an anastomotic stricture, twisted
descended pull-through colon, or rolling of a long muscular cuff left
behind. They are managed by sequential dilatations or redo pull-though
surgery. Soiling can be caused by damage sphincter function
(incontinence), abnormal sensation and pseudo-incontinence. Anorectal
manometry is indicated to determine the cause. Soiling not associated
with constipation is caused by true fecal incontinence and does not
improve with time. Thus, to preserve the anal canal and avoid sphincter
damage are of vital importance during the TAPT. Enterocolitis is
managed with bowel rest, colonic irrigations, and systemic antibiotics.
Most children have an excellent quality of life going into adulthood
after TAPT. Besides the management of HD, the TAPT can be utilized for
children born with rectal atresia, for severe chronic idiopathic
constipation associated with megarectosigmoid, idiopathic rectal
prolapse, and children with rectal prolapse after anorectal
malformation correction.
References:
1- De la Torre-Mondragon L, Ortega-Salgado JA: Transanal endorectal
pull-through for Hirschsprung's disease. J Pediatr Surg. 33(8):1283-6,
1998
2- De La Torre L, Langer JC: Transanal endorectal pull-through for Hirschsprung disease: technique,
controversies, pearls, pitfalls, and an organized approach to the
management of postoperative obstructive symptoms. Semin Pediatr Surg.
19(2):96-106, 2010
3- Langer JC: Laparoscopic and transanal pull-through for Hirschsprung disease. Semin Pediatr Surg. 21(4):283-90, 2012
4- Mao YZ, Tang ST, Li S: Duhamel operation vs. transanal endorectal pull-through procedure for
Hirschsprung disease: A systematic review and meta-analysis. J Pediatr Surg. 53(9):1710-1715, 2018
5- Miyano G, Takeda M, Koga H, et al: Hirschsprung's disease in the
laparoscopic transanal pull-through era: implications of age at surgery
and technical aspects. Pediatr Surg Int. 34(2):183-188, 2018
6- Fosby MV, Stensrud KJ, Bjornland K: Bowel function after transanal
endorectal pull-through for Hirschsprung's disease - does outcome
improve with time?. J Pediatr Surg 55: 2375, 2378, 2020
7- Beltman L, Roorda D, Backes M, Oosterlaan J, van Heurn LWE, Derikx
JPM: Risk factors for short-term complications graded by Clavien-Dindo
after transanal endorectal pull-through in patients with Hirschsprung
disease. J Pediatr Surg. 2021 Aug 1:S0022-3468(21)00533-9. doi:
10.1016/j.jpedsurg.2021.07.024.
8- Karlsen RA, Hoel AT, Fosby MV, et al: Comparison of clinical
outcomes after total transanal and laparoscopic assisted endorectal
pull-through in patients with rectosigmoid Hirschsprung disease.
J Pediatr Surg. 2022 Jan 15:S0022-3468(22)00046-X. doi: 10.1016
PSU Volume 58 NO 05 MAY 2022
Slipping Rib Syndrome
Slipping rib syndrome (SRS) is a rare cause of lower rib and
abdominal pain in children and adults. SRS accounts for approximately
5% of all musculoskeletal chest pain in primary care. SRS is caused by
a hypermobility of the anterior false ribs that allows the 8th to 10th
ribs to slip or click as the cartilaginous rib tip abuts under the rib
above. The costal cartilage slips out of its normal anatomical
position, the anterior false ribs (8th through 10th) slide out of
orientation and become pinned underneath their adjacent superior ribs.
This displacement is caused by a congenital anomaly, damage to the
fibrinous articulation, or hypermobility of unknown origin. Impingement
of the intercostal nerve along the adjacent surface of the adjacent
ribs during this slip occurs causing acute pain. Failure to diagnose
this condition might lead to unnecessary tests in the child. The pain
is caused when the lower costal cartilages 8th to 10th which are not
connected to the sternum lose their fibrous or cartilaginous
attachments to each other. Pain can extend to the floating ribs (11th
and 12th). Movements in the child such as twisting, simple lumbar
flexion, bending, deep breathing, laughing, sitting, sneezing, or
coughing results in irritation of the intercostal nerve hence pain.
Pain is sharp, intermittent, stabbing lasting for a few minutes
followed by a dull or burning sensation for several hours. Sometimes
associated with nausea and vomiting. Slippage may produce a clicking or
popping sensation. Pain can range anywhere from the midline to the
lateral flank and from the xiphoid process to as inferiorly as the
umbilical line. Due to interconnections between intercostal and somatic
visceral nerves the pain might be interpreted as upper abdominal
(subcostal). The lack of significant radiographic findings makes the
diagnosis of SRS difficult using imaging. Time to diagnosis is often
years. Differential diagnosis includes rib fracture, chondritis and
pleuritic pain. There is usually one dominant affected side, though the
syndrome can occur bilaterally. SRS can occur in any age group and
females athletes (swimmers) are more commonly affected than males. Many
of these patients have hypermobile joints with laxity and subluxation
without symptoms. Diagnosis is established through history and physical
examination as imaging are not very useful. Dynamic US can be of help
in the diagnosis by demonstrating an overlapping movement of the lower
rib above the upper rib. Less thickness of the ipsilateral rectus
abdominis muscle has also been found. Pain is intermittent and
localized to the lower ribcage with some trigger point of tenderness
palpable. Popping or grinding sensation with movement can be elicited.
At physical exam the hook maneuver, the examiner slides his finger
under the costal margin and lift anteriorly and superiorly, reproducing
a click and pain and diagnosing the condition. Careful palpation can
identify the disconnected cartilage, or the cartilage curling beneath
the overlying ribcage causing point tenderness. Compressing on the
sides of the ribs simultaneously may reproduce the pain at the affected
costal margin. As a temporary and localizing measure, a local
anesthetic rib block with Bupivacaine can be performed which provides
temporary or complete symptoms relief. Repeated anesthetics and steroid
rib block, manipulative techniques, acupuncture, Botox injections,
prolotherapy, and topical anesthetics produce long-lasting results.
Should conservative measures fail, the management should consist of
removing the slipping, disconnected cartilages through a small incision
at the affected lower subcostal margin. The hypermobile cartilage is
removed all the way to the costochondral junction in most cases
including the perichondrium to avoid regrowth of the cartilage. During
this maneuver, the intercostal neurovascular bundle is preserved.
Injury to the neurovascular bundle can cause acute blood loss or
chronic neuropathic pain. The use of a vertical bioabsorbable plate to
stabilize the hyperflexible subluxing bony ribs used to stabilize
fractures is also highly recommended to be incorporated with resection
of the cartilages as recurrence rate are decreased significantly. If
the patient develops recurrent symptoms after surgery, this might be
due to missed slipping cartilages during the initial procedure,
regrowth of the cartilage, or new symptoms on the contralateral side.
Most patients are satisfied after surgical resection of the slipping
ribs.
References:
1- McMahon LE: Slipping Rib Syndrome: A review of evaluation, diagnosis and treatment. Semin Pediatr Surg. 27(3):183-188, 2018
2- Gress K, Charipova K, Kassem H, et al: A Comprehensive Review of
Slipping Rib Syndrome: Treatment and Management. Psychopharmacol Bull.
50(4 Suppl 1):189-196, 2020
3- Fraser JA, Briggs KB, Svetanoff WJ, St. Peter SD: Long-term outcomes
and satisfaction rates after costal cartilage resection for slipping
rib syndrome. J Pediatr Surg 56: 2258-2262, 2021
4- Foley Davelaar CM: A Cinical Review of Slipping Rib Syndrome. Curr Sports Med Rep. 20(3):164-168, 2021
5- McMahon LE, Salevitz NA, Notrica DM: Vertical rib plating for the
treatment of slipping rib syndrome. J Pediatr Surg. 56(10):1852-1856,
2021
6- Romano R, Gavezzoli D, Gallazzi MS, Benvenuti MR: A new sign of the
slipping rib syndrome? Interact Cardiovasc Thorac Surg. 34(2):331-332,
2022
Encapsulating Peritoneal Sclerosis
Peritoneal dialysis is the preferred chronic dialysis modality in
the pediatric age. Encapsulating peritoneal sclerosis (EPS) is a rare
but most serious complications of continuous ambulatory peritoneal
dialysis (CAPD) in end-stage renal patients. The incidence of EPS in
children receiving CAPD is 2-15% depending on the long-term use of
CAPD, while the mortality exceeds 30% with most survivors requiring
long-term parenteral nutrition. EPS is characterized by progressive
fibrosis of the peritoneum resulting in reduced ultrafiltration
capacity, dysfunctional peristalsis of the bowel, and partial or
complete bowel luminal obstruction. EPS involves both the visceral and
parietal peritoneum. EPS is not specific to CAPD children and can be
seen secondary to drug therapy (Beta-blockers), sarcoidosis, systemic
lupus erythematosus, abdominal tuberculosis, gastrointestinal
malignancies, protein S deficiency and ovarian luteinized thecomas. EPS
is a clinical syndrome that continuously, intermittently, or repeatedly
presents with symptoms of intestinal obstruction due to adhesions of
diffusely thickened peritoneum. The diagnosis of EPS is based on
clinical, histologic, and radiographic findings. The pathophysiological
event in the development of EPS is an inflammatory process resulting in
loss of the mesothelial layer of the peritoneum and fibroconnective
tissue proliferation. Fibrin deposition and fibrinolysis with
hyalinization of the superficial stromal collagen possibly tanned
through nonenzymatic glycosylation by the dialysate glucose plays an
important role in causing excessive fibrogenesis in children with CAPD.
Contributing events include duration of CAPD (single most significant
risk factor), recurrent episodes of bacterial or fungal peritonitis,
the acetate dialysis solution, chlorhexidine, plasticizers, and
cumulative exposure to hypertonic glucose-based dialysis solutions. The
rate of developing EPS increases with the years receiving CAPD. If a
child receives CAPD for more than five years and shows poor
ultrafiltration with peritoneal calcifications on CT-Scan, a peritoneal
biopsy should be performed to rule out the presence of severe EPS.
Peritoneal biopsies of EPS show loss of normal mesothelial cells,
massive expansion of the submesothelial compact zone and increased
vascularization, mononuclear cell infiltration, calcifications, and low
peritoneal mast cell number. EPS occurs insidiously with vague
presenting symptoms initially. Symptoms of EPS include weight loss,
malnutrition, low-grade fever, nausea, vomiting, hemorrhagic effluent,
ultrafiltration failure, ascites, and recurrent bouts of abdominal pain
to subacute or acute intestinal obstruction with bowel necrosis.
Imaging might show dilated small bowel loops, air-fluid levels, and
calcific plaques. Ultrasound is the most sensitive modality to detect
EPS demonstrating thickened bowel wall with a trilaminar appearance and
adhesion of bowel loops to the anterior abdominal wall. CT-Scan
demonstrates peritoneal thickened, bowel teetering, thickened bowel
wall, loculated ascites, peritoneal calcification, and clouding of
mesenteric fat. The cocoon appearance is secondary to the presence of a
thick fibrous layer encapsulating the small bowel. Some recommendations
to stop peritoneal dialysis in cases with EPS include ultrafiltration
failure, bloody deacylate with calcifications of the peritoneum,
duration longer than eight years of dialysis, persistency elevated
C-reactive protein and recurrent peritonitis. EPS can occur up to five
years after withdrawal of CAPD, when on hemodialysis, or after
transplanted. These cases are all characterized by an acute
presentation with a rapid clinical course. Management of EPS includes
termination of peritoneal dialysis, immunosuppression, steroids
(anti-inflammatory), tamoxifen (antifibrotic agent), and surgical
debridement. Surgical therapy is required when the child does not
respond to medical therapy or presents with complete bowel obstruction,
bowel perforation or hemoperitoneum. Total enterolysis and Noble
plication are the methods suggested in the literature. Preventing EPS
includes minimizing dialysate glucose exposure, preventing acute
peritoneal dialysis peritonitis and using a neutral-pH solution.
References:
1- Honda M, Warady BA: Long-term peritoneal dialysis and encapsulating peritoneal sclerosis in
children. Pediatr Nephrol. 25(1):75-81, 2010
2- Debska-Slizien A, Konopa J, Januszgo-Giergielewicz B, et al:
Posttransplant encapsulating peritoneal sclerosis: presentation of
cases and review of the literature. J Nephrol. 26(5):906-11, 2013
3- Stefanidis CJ, Shroff R: Encapsulating peritoneal sclerosis in children. Pediatr Nephrol. 29(11):2093-103, 2014
4- Brown EA, Bargman J, van Biesen W, et al: Length of Time on
Peritoneal Dialysis and Encapsulating Peritoneal Sclerosis - Position
Paper for ISPD: 2017 Update. Perit Dial Int. 37(4):362-374, 2017
5- Jagirdar RM, Bozikas A, Zarogiannis SG, Bartosova M, Schmitt CP,
Liakopoulos V: Encapsulating Peritoneal Sclerosis: Pathophysiology and
Current Treatment Options. Int J Mol Sci. 20(22):5765, 2019
6- Sharma V, Moinuddin Z, Summers A, et al: Surgical management of
Encapsulating Peritoneal Sclerosis (EPS) in children: international
case series and literature review. Pediatr Nephrol. 2021 Aug 26. doi:
10.1007/s00467-021-05243-0.
7- Keese D, Schmedding A, Saalabian K, Lakshin G, Fiegel H, Rolle U:
Abdominal cocoon in children: A case report and review of literature.
World J Gastroenterol. 27(37):6332-6344, 2021
Burnout Syndrome
Burnout syndrome is a psychological syndrome arising from the
continued response to chronic interpersonal stressors while at work.
Defined as a state of physical and mental exhaustion related to
caregiving activities or work is a severe problem affecting medical
personal and healthcare organizations. Emotional exhaustion and
irritability in the work environment leads to development of
psychiatric problems characterized by emotional exhaustion,
depersonalization, and diminished personal accomplishment. The three
main components of burnout are overwhelming exhaustion, feeling of
cynicism or depersonalization, and a sense of ineffectiveness and lower
efficacy. Burnout is the most common mental issue faced by residents
and junior attending. The prevalence among residents is over 50%
comparable between medical and surgical specialties. Older age and male
prevalence, especially married male residents, are associated with
higher prevalence. Systematic review studies have revealed that women,
young surgeons, single status, increased workload, and conflict with
other colleagues is associated with a higher risk of burnout. In either
case, men manifested more depersonalization symptoms, whereas women
report more emotional exhaustion symptoms. Women are more likely to
adopt emotionally based strategies (self-blame, seeking comfort,
getting help and advice from others), while men often resort to
ego-defense mechanisms like depersonalization which might impact the
ability to provide care. The consequences of developing burnout
syndrome between physicians and residents in-training include physical
illness, increase feelings of hopeless, irritability, impatience poor
interpersonal relationships, risk of medical errors, reduced medical
service, depression, and adverse effect in patient safety. In the case
of residents, they are subjected to sleep deprivation, high workload,
unsatisfactory salaries, and high responsibility. Symptoms of burnout
can originate from causes such as: bureaucratic requirements, continued
changing work environment, micro-management by administrations, poor
clinical supervision, sensationalist media reports of medical errors,
limited healthcare resources, litigious environment, and poor work-life
balance. Residents and interns in urgency and surgical specialties such
as general surgery, anesthesiology, obstetrics & gynecology, and
orthopedics have the highest prevalence of burnout syndrome. A
plausible explanation is that residents in these specialties deal with
emergency routines dealing directly with life-threatening situations
with overload shifts. Specialties with less burnout syndrome include
otolaryngology, plastic surgery, and neurology. To deal with the
problem of burnout in residencies it is necessary to recognize that the
problem exists and describe its impact. By addressing burnout, you can
increase your personal wellness, improve patient safety, satisfaction,
and quality of life. Burnout syndrome needs evaluations of the
prevalence and intensity using validated instruments such as the
Maslach Burnout inventory and apply these to students, interns,
residents and attending periodically. The Maslach burnout inventory
toolkit asses the level of burnout by measuring: 1) emotional
exhaustion caused by work, 2) depersonalization, translated into
unfeeling and impersonal response toward recipients of our service,
care or treatment, and 3) personal accomplishment, measure by the
feeling of competence and successful achievement in our work toward
patients. In addition, the work environments should be asses by using
the Areas of Worklife Survey which measures workload (amount of work to
be done), control (opportunity to make choices and decisions to solve
problems), reward (recognition, financial and social you receive for
contributing into the job), community (quality of the social context in
which you work), fairness (extent to which organizations has
consistency and equitable rules for everyone) and values (what matters
to you in your work). Burnout is prevalent in more than half of US
physicians, makes us less happy with our professional choice and less
effective in our roles as clinicians, teachers, mentors, and role
models. Neglected burnout leads to alcohol and substance misuse,
anxiety, depression, discontinuation of residency, fatigue, impaired
interpersonal and marital relationship, insomnia and even suicide.
Working more than 60-80 hours per week and taking two or more nights on
call per week promote burnout. Managing burnout syndrome can be divided
into preventive and therapeutic measures. Actions must be concentrated
on risk factors, improvement in the relationship between professionals
and the promotion of healthy behavior in physicians. Therapeutic
strategies consist in management of negative emotions and relaxation
technique. To alleviate and prevent burnout we should fix what we
control and advocate for the rest. These include aligning with patients
providing high-quality care, develop expertise in disease and not
technique, redesign the practice, developing progressive employment
policies, and respect toward our own choices.
References:
1- Rodrigues H, Cobucci R, Oliveira A, et al: Burnout syndrome among
medical residents: A systematic review and meta-analysis. PLoS One.
2018 Nov 12;13(11):e0206840. doi: 10.1371/journal.pone.0206840.
eCollection 2018.
2- Doherty GM: How Do We Prevent Burnout in Surgery? Adv Surg. 53:131-143, 2019
3- Low ZX, Yeo KA, Sharma VK, et al: Prevalence of Burnout in Medical
and Surgical Residents: A Meta-Analysis. Int J Environ Res Public
Health. 16(9):1479, 2019
4- Bratu I, Heiss K, Mueller C, Winthrop A, Blair G, Moulton CA:
Canadian Asssociation of Pediatric Surgeon's state of wellness. J
Pediatr Surg. 54: 891-894, 2019
5- Galaiya R, Kinross J, Arulampalam T: Factors associated with burnout
syndrome in surgeons: a systematic review. Ann R Coll Surg Engl.
102(6):401-407, 2020
6- Huang R, Hewitt DB, Cheung E, et al: Burnout Phenotypes Among U.S.
General Surgery Residents. J Surg Educ. 78(6):1814-1824, 2021
PSU Volume 58 NO 06 JUNE 2022
DIC
Disseminated intravascular coagulation (DIC) is a state of
hemostatic dysregulation which causes microvascular clotting and
consumptive coagulopathy and is seen in a variety of conditions
including sepsis (most common), trauma, malignancy, liver diseases, and
toxins. These conditions can cause cytokine-induced endothelial and
mononuclear cell release of tissue factors and generation of excessive
thrombin extending outside the local area of injury along with release
of fibrinolytic proteins. Excessive thrombi production leads to
microvascular thrombi with consumption of platelets, procoagulant and
anticoagulants proteins, and inhibition of fibrinolysis. All these
factors contribute to multiorgan failure. Diagnosing DIC is difficult
since it includes a wide range of clinical presentations, including
mild to excessive bleeding and systemic thromboembolic phenomenon
associated with multiorgan failure. The International Society on
Thrombosis and Hemostasis (ISTH) diagnostic scoring system for overt
DIC is widely utilized in intensive care units for diagnosing DIC,
including sequential testing of components of the ISTH scoring system
such as prothrombin time, platelet count, fibrinogen, and D-dimer.
Fibrinogen does not seem to have a significant impact on the prediction
of DIC. Among individual DIC components evaluated, prolonged PT is the
most predictive of increased vasopressor use, followed by elevated
D-dimer. Pediatric patients presenting with suspected sepsis to the
emergency department who have scores greater than three are more likely
to have outcomes including increased vasopressor use, increase
mortality, prolonged hospital and intensive care unit lengths of stay,
increased rates of mechanical ventilation, and increase mortality. The
predominant condition leading to DIC is sepsis affecting 75,000
children per year in the USA with mortality rates ranging between 7010%
among all age groups. Sepsis is a syndrome with a variety of clinical
manifestations including organ dysfunction caused by a dysregulated
host response to the inciting infection. Septic shock produces profound
circulatory, cellular, and metabolic abnormalities associated with
high-risk morbidity and mortality. Coagulation activation can be
triggered with septic shock leading to activation and inhibition of
physiological anticoagulation mechanisms and fibrinolytic system
leading to intravascular fibrin formation and consumption of
procoagulant leading to DIC. DIC then results in hemorrhage or
thrombotic occlusion of vessels leading to inadequate blood and oxygen
to various organs leading to multiple organ dysfunction syndrome. DIC
results from the acceleration of the clotting cascade, inactivation of
endogenous anticoagulants, and modification of fibrinolysis, leading to
hypercoagulability and augmented fibrinolysis. This event causes the
formation of multiple microthrombi in the systemic circulation, which
consequently manifests as multiorgan failure. The main management of
DIC is early identification and treatment of the underlying condition,
hemodynamic support, frequent monitoring of laboratory and clinical
parameters, and replacement of consumed coagulation factors and blood
components via transfusion of platelets, fresh frozen plasma, or
cryoprecipitate. Prophylactic transfusion of these blood products is
not recommended unless there is clinical bleeding or impending invasive
procedure (surgery). There is a demonstrated efficacy of antithrombin
and protein C concentrates, recombinant activated protein and
recombinant thrombomodulin for the management of DIC in children.
Activated protein C inactivates coagulation factors V and VIII, and
ultimately causes inhibition of thrombin formation and has
anti-inflammatory properties. Recombinant thrombomodulin activates
protein C leading to the inactivation of factor Va, which ultimately
leads to the inhibition of thrombin generation.
References:
1- Soundar EP, Jariwala P, Nguyen TC, Eldin KW, Teruya J: Evaluation of
the International Society on Thrombosis and Haemostasis and
institutional diagnostic criteria of disseminated intravascular
coagulation in pediatric patients. Am J Clin Pathol. 139(6):812-6, 2013
2- Jhang WK, Ha E, Park SJ: Evaluation of disseminated intravascular
coagulation scores in critically ill pediatric patients with septic
shock. J Crit Care. 47:104-108, 2018
3- Slatnick LR, Thornhill D, Deakyne Davies SJ, et al: Disseminated
Intravascular Coagulation Is an Independent Predictor of Adverse
Outcomes in Children in the Emergency Department with Suspected Sepsis.
J Pediatr. 225:198-206.e2, 2020
4- Rajagopal R, Thachil J, Monagle P: Disseminated intravascular coagulation in pediatrics. Arch Dis Child. 102: 187-93, 2017
5- Kunwar S, Alam M, Ezekwueme F, et al: Diagnostic Scores and
Treatment Options for Acute Disseminated Intravascular Coagulation in
Children. Cureus. 13(9):e17682, 2021
6- Jhang WK, Park SJ: Evaluation of Disseminated Intravascular
Coagulation in Critically Ill Pediatric Hemato-oncology Patients with
Septic Shock. Thromb Haemost. 120(11):1505-1511, 2020
Extracolonic Manifestations of FAP
Familial adenomatous polyposis (FAP) is a hereditary
syndrome of autosomal dominant inheritance caused by a germline
mutation in the adenomatous polyposis coli (APC) tumor suppressor gene
in the long arm of chromosome 5 characterized by 100-1000 adenomatous
polyps in the colon and rectum with progression to cancer if left
untreated. The less aggressive variant termed attenuated FAP exhibits
fewer colorectal adenomatous polyps (10-100), later age of adenoma
appearance and a lower cancer risk. FAP accounts for 1% of all
colorectal cancers in the USA. Many of these hereditary syndromes have
extracolonic manifestations, including the development of benign and
malignant tumors. These extracolonic manifestations can be the first
sign of the inherited syndrome helping establish an early diagnosis of
FAP in high-risk patients who have not yet developed colorectal
polyposis. Extracolonic cancer in FAP has become of significant concern
since affected patients are living longer and are increasingly being
diagnosed earlier. The majority of FAP patients (over 70%) present with
some level of extracolonic manifestation during the course of the
disease. Extracolonic manifestation can be classified according to the
tissue of origin into ectodermal, endodermal, and mesodermal.
Ectodermal lesions include epidermoid or sebaceous cysts which occur
with increasing frequency at a younger age with predilection in the
face, scalp, legs, and arms. Also, congenital hypertrophy of the
retinal pigment is a well-recognized ocular sign and clinical marker
occurring in more than 90% of FAP patients. Ocular lesions are
discrete, darkly pigmented, round, oval or kidney shape raging in size
from 0.1-1.0 optic disk diameter. Lesions of mesodermal origin include
desmoid disease, lipomas, fibromas, osteomas, and dental abnormalities
such as odontomas, dentigerous cysts and supernumerary teeth. Dentists
should consider referral of patients with extranumerary teeth or jaw
osteomas for FAP evaluation. Desmoid tumors are slow growing
mesenchymal neoplasms composed of fibroblasts and myofibroblasts within
a rich collagen matrix. Desmoid tumors are most commonly (80%)
intraabdominal (mesentery of retroperitoneum). Other sites include
abdominal wall, subcutaneous, and in the musculo-aponeurotic layer.
They can reach massive size, cause intestinal obstruction, mesenteric
vascular obstruction, and ureteric obstruction. They lack a metastatic
potential but exhibit aggressive local behavior with infiltrative
patterns of growth involving surrounding structures and a high local
recurrence rate following resection. Desmoid tumor development can be
exacerbated by surgical trauma or pregnancy. The frequency of
developing desmoid tumors is higher for FAP with a lifetime risk of 8%
for males and 15% for females. Management of desmoid tumors is complex
and depends on location, symptoms, extent of disease and pattern of
growth. Radiation therapy, NSAID's, antiestrogen and chemotherapeutic
are options. Surgery is used for those unresponsive to medical
treatment or if complications requiring emergency surgery occur.
Recently, Imatinib has been shown to positively impact progression-free
survival in patients with advance desmoids. Osteomas may occur in the
mandible, maxilla, sinuses or calvarium of the skull. Exostosis may be
found in the skull, digits, and long bones. Lesions of endodermal
origin include gastrointestinal adenomas and carcinomas. After the
colon and rectum, the duodenum is the second most common site of polyp
development in patients with FAP. Non-adenomatous fundic gland polyps
predominate in the stomach, while duodenal lesions were mostly
adenomatous in nature. The most common extracolonic manifestations with
FAP are upper gastrointestinal polyps, including gastroduodenal
adenomas that can progress to cancer. Upper GI polyps in the setting of
FAP are located in the stomach, duodenum and periampullary region.
Gastric polyps are typically non-adenomatous benign fundic gland polyps
considered hamartomas. They develop in almost 50% of FAP patients, are
located in the antrum, and are not associated with cancer. Duodenal
adenomatous polyps in FAP are found in 30-70% of individuals, have a
predilection for the second and third portion of the duodenum, have a
strong propensity toward developing into duodenal cancer and a
genotypic/phenotypic correlation with mutation in exon 15 of the APC
gene has been described. Duodenal cancer is the second most common
cause of disease-related mortality in patients with FAP with a lifetime
risk of approximately 3-5%. Is one of the leading causes of death in
patients who have undergone prophylactic colectomy. Increasing age
appears to be correlated with higher risk of progression to advanced
polyposis. Endoscopic surveillance is recommended starting between age
25 and 30 or 5 years earlier than any affected family member with
duodenal adenomatous polyps. The Spigelman classification is used to
evaluate polyp severity based on number, size, histology, and presence
of dysplasia. Management of duodenal polyps consists of pharmacologic
(non-steroidal inflammatory drugs) which has been found ambiguous,
endoscopic, or surgical removal. Among FAP patients the risk of death
is higher than in the general population (3-fold); disease related
mortality is caused more commonly by upper gastrointestinal
malignancies followed by desmoid tumors and perioperative
complications. Other less common extraintestinal malignancies
associated with FAP include thyroid, brain (medulloblastoma), adrenal,
hepatoblastoma and pancreatobiliary tumors. Thyroid cancer is the third
most common malignancy associated with FAP. Is most commonly papillary
histology, presents in the second or third decades of life, mostly in
females. The cribriform-morular histologic variant of papillary thyroid
cancer is highly suggestive of FAP. Periodic thyroid ultrasound
screening should be considered in patients with FAP, and FNA in those
with thyroid nodules. The risk of pancreatic adenocarcinoma is also
increased in patients with FAP. Mutations in the APC gene are
associated with several other extraintestinal manifestations including
Turcot and Gardner syndromes. Gardner's syndrome is characterized by
the typical manifestations of FAP and presence of osteomas, fibromas,
and epidermoid cysts.
References:
1- Anaya DA, Chang GJ, Rodriguez-Bigas MA: Extracolonic manifestations
of hereditary colorectal cancer syndromes. Clin Colon Rectal Surg.
21(4):263-72, 2008
2- Campos FG: Surgical treatment of familial adenomatous polyposis:
dilemmas and current recommendations. World J Gastroenterol.
20(44):16620-9, 2014
3- Kennedy RD, Potter DD, Moirt CR, El-Youssef M: The natural history
of familial adenomatous polyposis syndrome: A 24 year review of a
single center experience in screening, diagnosis, and outcomes. J
Pediatr Surg. 49: 82-86, 2014
4- Campos FG, Sulbaran M, Safatle-Ribeiro AV, Martinez CA: Duodenal
adenoma surveillance in patients with familial adenomatous polyposis.
World J Gastrointest Endosc. 7(10):950-9, 2015
5- Mitchem JB, Hall JF: Adenomatous Polyposis Syndromes: Diagnosis and Management. Clin Colon Rectal Surg. 29(4):321-329, 2016
6- Besteiro B, Gomes F, Costa C, Portugal R, Garrido I, Almeida J:
Importance of Extraintestinal Manifestations in Early Diagnosis of
Gardner Syndrome. Case Rep Gastrointest Med. 2020 Aug 4;2020:7394928.
doi: 10.1155/2020/7394928. eCollection 2020
Delayed Separation Umbilical Cord
The umbilical cord connects the fetus to the placenta in the
uterus, is made of blood vessels (two arteries and one vein), and
connective tissue. The umbilical cord of newborn infants separates
during the first two weeks of life, though there is a wide variation in
the time this event takes place with regard to ethnicity, geographical
location and proper care of the cord. The mean umbilical separation
time is 6.6 days, with a median of seven days. The cord stump dries and
falls off, and the wound heals. The cord separation is mediated by
leukocytes. Histologically this process is characterized by granulocyte
influx and phagocytosis at the base of the cord. Delayed separation of
the umbilical cord is defined as separation that occurs after three
weeks of life. Factors associated with delayed separation of the cord
include the use of alcohol or chlorhexidine for cleaning purpose,
prematurity, and infants born by cesarean section. Neonates delivered
by cesarean section tend to have longer cord separation time due to
less bacterial colonization after birth. The use of postpartum
antibiotics, parenteral nutrition and phototherapy also delayed the
separation of the umbilical cord. Sepsis delays the cord separation
time by sixfold beyond the second week of life. Urachal anomalies
rarely can be associated with delayed separation. According to the
National Institute of Health Care and Excellence guidelines, parents
should be advised how to keep the umbilical cord clean and dry, and
that antiseptics should not be used routinely. A marked delayed in
umbilical cord separation raises the suspicion of leukocyte adhesion
deficiency (LAD), a rare autosomal recessive hereditary disorder
leading to defective neutrophil function. LAD is a disorder of
neutrophils due to a genetic defect in the beta subunit of the integrin
molecule ITGB2 which encodes the integrin beta chain-2 protein CD18.
This defect leads to dysfunction of leukocyte adhesion to the wall of
blood vessels and migration of leukocytes to sites of infection and
inflammation. Patients have a complete absence of neutrophils at the
site of inflammation causing recurrent bacterial infections and sepsis.
LAD is characterized by recurrent infections, impaired pus formation,
delayed wound healing, omphalitis, and delayed separation of the
umbilical cord as hallmark features of the disease. Patients are
infected with common pathogenic agents but not opportunistic ones and
respond well to antimicrobial therapy. Proteus, Klebsiella,
Staphylococcus aureus, Pseudomonas aeruginosa, and enterococci are the
most common pathogens affecting LAD patients. The severe form of this
disease remains a life-threatening condition with limited 2-years
survival in the absence of transplantation. The flow cytometric
analysis of monocytic intracellular tumor necrosis factor-alpha
production in response to lipopolysaccharide may be a useful method to
screen for the disease. Management of LAD consists of allogeneic stem
cell or bone marrow transplantation.
References:
1- Lopez-Medina MD, Lopez-Araque AB, Linares-Abad M, Lopez-Medina ML:
Umbilical cord separation time, predictors and healing complications in
newborns with dry care. Plos ONE 15(1): e0227209, 2020
2- Aygun C, Subasi A, Kucukoduk S: Timing of umbilical cord separation
and neonatal intensive care unit practices. Am J Perinatol. 22(5):
249-251, 2005
3- Bouhouche A, Tabache Y, Askander O, et al: Novel ITGB2 Mutation Is
Responsible for a Severe Form of Leucocyte Adhesion Deficiency Type 1.
Biomed Res Int., doi: 10.1155/2022/1141280. 2022
4- Unal S, Demirel N, Arslan Z, Tokgoz-Cuni B, Ulubas-Isik D, Bas AY:
Umbilical Cord Separation Time and Influencing Factors in
Very-Low-Birth-Weight Preterm Neonates. Am J Perinatol. 2021 Mar 3.
doi: 10.1055/s-0041-1726035.
5- Takada H, Yoshikawa H, Imaizumi M, et al: Delayed separation of the
umbilical cord in two siblings with Interleukin-1 receptor-associated
kinase 4 deficiency: rapid screening by flow cytometer. J Pediatr.
148(4):546-8, 2006